 |
|
|
| |
| |
High Dose Peginterferon for Non-responders, and Naïve & Relapsers
|
|
| |
| |
Reported by Jules Levin
I think you'll see from the preliminary study data that it appears as if high dose peginterferon can increase viral clearance in IFN/RBV nonresponders compared to standard doses of PEG/RBV. But the problem may be tolerability. The Pegasys high dose study results are early data but suggest better responses with high dose Pegasys. The PegIntron data below reports week 24, 48, and 72 week preliminary data and supports better viral clearance and tolerability concerns after week 24. However, for previous nonresponders to IFN/RBV using the higher Peginterferon dose in combination with RBV preliminarily appears to be a feasible strategy based on the data we have so far that might yield improved viral response rates compared to using standard Peginterferon dose if you can tolerate the higher doses. Waiting for final study results will provide more comprehensive information for making treatment decisions for nonresponders.
High Dose Pegasys plus Ribavirin in Patients with Genotype 1/HCV Who Failed Interferon/ribavirin
The objective of this study was to determine whether treatment with higher than usual doses of Pegasys combined with ribavirin (Copegus) improves virologic response rates after 12 weeks of treatment in patients with chronic hepatitis C with HCV genotype1, who did not respond to previous therapy with conventional interferon plus ribavirin. Doses of Pegasys used in the study were 180 mcg/wk, 270 mcg/wk, and 360 mcg/wk. After 12 weeks patients receiving the high dose Pegasys achieved superior response rates and tolerability, adverse events and discontinuations was the same across all 3 dose regimens. The study is ongoing and we need to wait for further data at 48 and 72 weeks to better evaluate the high dose approach.
Patients were >18 yrs old and had not responded to >22 weeks of treatment with interferon plus RBV. This study is randomized, open-label, and was conducted at multiple centers. Patients were required to have HCV RNA levels >1000 IU/ml (Cobas Amplicor HCV Monitor Test v2.0), persistently elevated ALT for >6 months, and a liver biopsy obtained within the previous 36 months.
Patients were excluded if they had hepatitis A or B, or HIV, decompensated liver disease, cirrhosis, or chronic liver disease other than chronic HCV.
Other exclusion criteria included hemoglobin <12 g/dl in women or <13 g/dl in men; neutrophil count <1,500 cells/mm3 or platelet count <90,000 cells/mm3; serum creatinine level >1.5 times theupper limit of normal; severs psychiatric disease; severe seizure disorder or current anticonvolsant use; immunologically mediated disease; chronic pulmonary disease with functional limitation; severe cardiac disease; poorly controlled thyroid disease; severe retinopathy; and major organ transplantation.
The study provided 48 weeks treatment plus a 24 week untreated follow-up period. Patients were randomized to treatment with Pegasys 180 ug/wk, 270 ug/wk, or 360 ug/wk for 12 weeks, after which all patients receiveda dosage of 180 ug/wk for a further 36 weeks. All patients received ribavirin for the full 48 weeks (1000 mg/day for patients weighing <75 kg; 1200 mg/day for those weighing >75 kg). The dose of Pegasys could be reduced in a stepped fashion in the event of neutropenia, thrombocytopenia, or elevated ALT. The lowest allowable dose was 45 ug/wk. The dose of RBV could be reduced to manage anemia.
Patients who failed to achieve an early virologic response, defined as undetectable HCV RNA (<50 IU/ml by qualitative PCR) after 12 weeks, discontinued treatment.
The primary efficacy variable was the early virologic response, defined as undetectable HCV RNA by qualitative PCR (Cobas Amplicor HCV Test v2.0) after 12 weeks of combination therapy with Pegasys plus ribavirin.
RESULTS
A total of 72 patients were randomized and received treatment. 75% of patients in 180 and 270 arms were men, 83% in 360 arm were men. Age was 40-44 across all three arms. Weight was 75 kg in 180 and 270 arms, 80 kg in 360 arm. BMI was 26-27 kg/m2 across all 3 arms.
Early virologic responses after 12 weeks of treatment were obtained in 21%, 35%, and 45% in the low (180 ug/wk), intermediate (270 ug/wk), and high dose groups (360 ug/wk), respectively.
Virologic response rates were higher in patients with low viral load (<800,000 IU/ml) than high viral loads. Consistent with the overall results, the highest virologic response rates in patients with high and low viral loads were obtained in the high-dose group (360 ug/wk).
180 ug/wk dose: 26.7% in low viral load (n=15) vs 15.4% with low viral load (n=13) had early viral response
270 ug/week: 46% with low viral load (n=13) vs 14% with high viral load (n=7) had early viral response
360 ug/wk: 54% with low viral load (n=13) vs 36% with high viral load (n=11) had early viral response
TOLERABILITY
The study authors reported Pegasys was well tolerated during the 12 week study. The mean weekly dose of Pegasys was ≥96% of the dose prescribed dose in the randomization scheme. Requirements for dosage reductions did not increase in proportion to the dose of Pegasys.
Doses of Pegasys Administered
|
|
| |
| |
| |
180 ug/wk |
270 ug/wk |
360 ug/wk |
| |
n=28 |
n=20 |
1n=24 |
| Mean dose ug/wk |
172 |
263 |
349 |
| Mean total dose |
2121 |
3290 |
4368 |
| Dose reductions for
adverse events |
9(32%) |
2(10%) |
3(12.5%) |
|
|
| |
| |
The incidence of adverse events, severe adverse events, and lab abnormalities (including neutropenia and thrombocytopenia) were similar in the 3 treatment groups. Asthenia (40%), headache (34%), fever (31%), and myalgia (27%), were the most frequently reported adverse events. At week 12, grade 3 neutropenia was detected in 8 (29%), 5 (27%), and 3 (13%) patients treated with Pegasys 180 ug/wk, 270 ug/wk, and 360 ug/wk, respectively.
Adverse Events Reported During the First 12 Weeks of Treatment
|
|
| |
| |
| |
180 ug/wk |
270 ug/wk |
360 ug/wk |
| |
n=28 |
n=20 |
1n=24 |
| Patients w/AEs |
89% |
90% |
91% |
| Patients w/severe AE |
25% |
25% |
25% |
| Dose reductions for
adverse events |
- |
- |
4% |
|
|
| |
| |
Sustained virologic response rates and long-term safety data from the maintenance phase of the study will provide important informationabout high dose combination therapy.
High Dose PegIntron plus ribavirin for Prior Non-responders with HCV
John Gross and colleagues reported results from the RENEW study. We have seen disappointing results with PEG/interferon/ribavirin for IFN/ribavirin npn-responders. 10-11% of interferon/ribavirin non-responders treated with conventional doses of peginterferon + ribavirin have a sustained virological response. The current trial was designed to see if better results could be achieved by retreating with higher doses of peginterferon alfa-2b (PEG2b) + weight-based ribavirin.
The study goal was to compare the efficacy, safety and tolerability of three different doses of PegIntron alfa-2b + weight-based ribavirin among interferon/ribavirin non-responders. Patients were randomized to 1 yr of treatment with one of three PegIntron dose regimens:
PegIntron alfa-2b 0.5 ug/kg/wk
PegIntron alfa-2b 1.5 ug/kg/wk
PegIntron alfa-2b 3.0 ug/kg/week
plus ribavirin 12-15 mg/kg/day.
STUDY ANALYSIS
The study endpoints were:
--efficacy: sustained virologic response (SVR)
--safety/tolerability: rates f dose reduction and discontinuation
Sample size:
--estimates of SVR (1,5 vs 3.0): 12% vs 19%
--sample size to detect this difference
--90% power: 560/group (total 1120)
--80%: 418/group (total 836)
Treatment assignment was stratified for sex, race, HCV genotype and histologic fibrosis. Treatment was stopped at 24 wk if PCR(+). Doses were reduced by 33% for toxicity; growth factors were not allowed.
Enrollment took place between February 2001 and November 30, 2002. The anticipated final anaklysis is for May 31, 2004. 963 patients from 100 centers were enrolled. Enrollment was stopped in the low-dose group after FDA approval of higher doses of PegIntron alfa-2b. Follow-up is 80% complete. The study population is 31% female, 16% African-American, 93% genotype 1, and 63% stage 2/3/4. Average age: 47 yrs. Average BMI: 29.
RESULTS
VIRAL CLEARANCE RATES (% HCV RNA negative)
|
|
| |
| |
| |
0.5 mcg/kg |
1.5 mcg/kg |
3.0 mcg/kg |
| Week 24 |
16% |
30% |
39% |
| Week 48 |
10% |
19% |
20% |
| Wk 72 SVR |
4% |
7% |
11% |
|
|
| |
| |
| |
Relapse rate appears lower with 3.0 dose compared to 1.5 dose.
The authors commented: On-treatment virological response rates were dose-related at 24 weeks but less so at 48 weeks. This was partly due to a higher rate of discontinuation after a satisfactory response at 24 wk on the higher dose. (see discontinuation rates below).
Response Rate is Higher With Minimal Fibrosis |
|
| |
| |
| |
F0/1 |
F2/3 |
| 24 weeks |
37% |
29% |
| 48 weeks |
26% |
18% |
|
|
| |
| |
Response Rates According to Genotype and Fibrosis
Of note, as you can see genotype 2/3 with low fibrosis had a sustained viral response from week 24 to week 48. But genotypes 2/3 with more advanced disease (F2/3/4) lost viral response from 56% at week 24 to 21% at week 48.
Week 24
Geno 1-F0/1: 35%
Geno 1-F2/3/4: 25%
Geno 2/3-F0/1: 57%
Geno 2/3-F2/3/4: 56%
Week 48
Geno 1-F0/1: 22%
Geno 1-F2/3/4: 20%
Geno 2/3-F0/1: 53%
Geno 2/3-F2/3/4: 21%
Response Rate is Lower Among African Americans
|
|
| |
| |
| |
African Americans |
Others |
| week 24 |
20% |
34% |
| week 48 |
8% |
20% |
|
|
| |
| |
| |
Response Tate is Independent of BMI |
|
| |
| |
| |
BMI > Median |
BMI < Median |
| week 48 |
8% |
20% |
|
|
| |
| |
Overall Rates of Neutropenia
--24% for 0.5 dose
--31% for 1.5 dose
--35% for 3.0 dose
Neutropenia Less than 750
(million/L)
--8% for 0.5 dose
--10% for 1.5 dose
--11% for 3.0 dose
Rates of Dose Reduction
--32% for 0.5 mcg/kg dose
--33% for 1.5 mcg/kg
--45% for 3.0 mcg/kg
Premature Discontinuation
--for 0.5 dose: 22% overall disct and 7% due to adverse events
--for 1.5 dose: 27% overall disct and 12% due to adverse events
--for 3.0 dose: 27% overall disct and 12% due to adverse event
Most Common Adverse Events
|
|
| |
| |
| Event |
0.5 |
1.5 |
3.0 |
| Fatigue |
47% |
54% |
54% |
| Fever |
26% |
25% |
29% |
| Headache |
23% |
30% |
32% |
| Leukopenia |
12% |
21% |
23% |
| Neutropenia |
24% |
31% |
35% |
|
|
| |
| |
Serious Adverse Events
0.5 ug/kg 8
1.5 ug/kg 22
3.0 ug/kg 24
Dyspnea (shortness of breath) 10
Elective surgery 8
Chest pain 4
Neutropenia 4
Suicidal thoughts 4
Suicide attempt 2
Abdominal pain retinal bleed or tear 2
Rectal bleeding 2
DKA 2
The authors concluded:
--30% to 40% of interferon/ribavirin non-responders achieved initial clearance of viremia on peginterferon alfa-2b + ribavirin.
--Doubling the PegIntron alfa-2b dose to 3.0 mcg/kg/wk resulted in a higher viral clearance rate at 24 wk but a similar rate at 48 wk due in part to a higher rate of dose reduction after week 24
--With use of growth factors and strong support to minimiz dose reductions in the second 6 months, PEG/interferon plus ribavirin is a reasonable re-treatment strategy
--The higher dose of PegIntron alfa-2b 3.0 mcg/kg/wk was well tolerated, with a higher rate of dose reduction but an identical rate of discontinuation.
--There was no serious difference in safety or tolerability between 1.5 and 3.0 mg/kg
--Predictors of response were similar to those previously observed for treatment-naïve patients.
Double Dose of PegIntron for Naives, Nonresponders and Relapsers: interim 12 week results
INTERIM RESULTS OF A RANDOMIZED TRIAL OF 1.5µG/KG VS. 3.0 µG/KG PEGYLATED INTERFERON ALFA 2-B PEG-INTRON(R) (PEG) PLUS RIBAVIRIN (RBV) FOR NAïVE CHRONIC HEPATITIS C PATIENTS AND 3.0µG/KG PEG PLUS RBV FOR NON-RESPONDERS (NR) AND RELAPSERS (R) TO PREVIOUS THERAPY. (THE TARGET TRIAL).
Calvin L White (University of Texas Southwestern, Dallas, TX) presented the poster at AASLD.
Peg plus RBV is the mainstay of chronic hepatitis C treatment. However, an upper limit in terms of safety and efficacy for dosing with Peg has not been evaluated. They studied a group of naïve patients using either conventional weight-based dosing of Peg; (1.5 ug/kg) or double the dose (3.0 ug/kg) plus RBV 13 ± 2 mg/kg/day for 48 weeks.
Genotype 1 patients were randomized 1:1 and to receive medication accordingly in unblinded fashion, with intent to enroll a total of 1,100 naïve patients N, an additional 300 previous NR or R to any type of interferon + RBV in a non randomized arm, using only the 3.0 ug/kg Peg dose. Values for HCV RNA at week 12 were compared to those obtained at baseline.
RESULTS
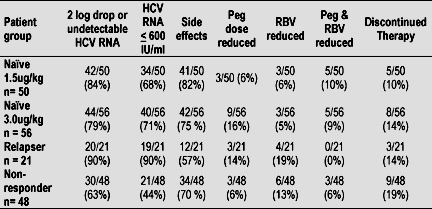
To date, 573 patients 306 N, 193 NR, 74 R have enrolled in the study. 175 patients 106 N, 48 NR, 21 R have reached week 12. HCV RNA results at week 12 are shown in the table below.
Treatment was well tolerated in most patients. Dose reductions were required in 20% of N 1.5ug/kg Peg and 29% of N 3.0ug/kg Peg patients. Side effects were equivalent between the two groups as shown in the table below.
Serious adverse events (SAE's) were less than 5% and equally observed in both arms of the N patients. None were directly attributed to study drug.
There were 25 patients in the wk 12 analysis that discontinued therapy. The reasons these patients were discontinued after reaching wk 12 were side effects (36%), +HCV RNA (32%).
It is interesting to note that 40% of the patients that were discontinued had > 2 log drop or negative HCV-RNA at week 12 and a positive HCV RNA was not a reason for their discontinuation.
The authors concluded that 3.0 ug/kg Peg dosing does not appear to improve 12 wk viral response, but we await 24 wk results and sustained viral response rates. Increased side effects as a result of doubling the interferon dose were not observed. More data is needed to verify the long-term efficacy and safety of this dosing regimen for patients with chronic hepatitis C.
This study was supported by a grant from Integrated Therapeutics Group a subsidiary of Schering Plough.
|
|
| |
 |
| |
|
|
|
|
|