 |
|
|
| |
|
Procrit for Ribavirin Induced Anemia: final study results
|
| |
| |
erage increased hemoglob
"Epoetin Alfa Treatment of Anemic HCV-Infected Patients Allows for Maintenance of Ribavirin Dose, Increases Meoglobin levels, and Improves Quality of Life vs Placebo: a randomized, bouble-blind, multicenter study"
Nezam Afdhal reported in an oral presentation at the DDW Conference (May 17-22, Orlando, FL) for the study research group investigating if EPO could benefit patients experiencing anemia due to IFN/RBV therapy. These are the final study results, and confirm previously reported interim results. The study examined HCV+ individuals but did not include HCV/HIV coinfected indivuduals, so perhaps a study in HIV-infected may be in order.in levels, and improved quality of life and less fatigue. BV) plus interferon, interfe exhibited on average increased hemoglobin levels, and improved quality of life and less fatigue.
Combination therapy for chronic HCV infection, ribavirin (RBV) plus interferon, interferon, or RBV/pegylated IFN (PEG-IFN), can induce anemia that may compromise the ability to achieve a sustained virologic response (SVR) by prompting RBV dose reduction or cessation. Treatment adherence is critical for achieving an SVR, and studies suggest that maintaining higher RBV doses (1000/1200 mg/day vs 800 mg/day) is more effective (Hadziyannis et al., EASL. 2002; Manns et al., Lancet. 2001; McHutchison, Hepatology. 2000). Results from studies suggest that particularly during early stages after starting treatment reduction in ribavirin dose may have crucial impact on viral load response by week 12 which in turn influences the capacity to achieve a sustained viral response. This study assessed whether epoetin alfa (EPO aka Procrit) treatment of anemic (hemoglobin £ 12 g/dL) HCV-infected patients could maintain RBV dose, increase hemoglobin (Hb) levels, and improve quality of life (QOL).
Data presented by Mitch Shiffman from the HALT-C study, which Afdhal referred to in this oral talk at DDW, showed dose reductions in ribavirin had a more negative effect than a dose reduction in Pegasys on end-of-treatment response and sustained viral response in the HALT-C study. Shiffman reported that sustained viral response was 23% (n=118, no dose reductions) and when there was a dose reduction only in Pegasys the SVR was still 23% (n=70). But when only ribavirin dose was reduced (n=36) the sustained viral response (SVR) was 11% (23% vs 11%, p<0.008). When there were dose reductions for both Pegasys and ribavirin the SVR was 8%.
This study is a double-blind, randomized, placebo-controlled study to assess, in anemic HCV-infected patients on combination RBV/interferon or RBV/Peg-IFN, the effect of once weekly epoetin alfa (EPO). This study was not designed to evaluate the effects of EPO on SVR.
HCV patients (N=186) who developed anemia (Hb <12 g/dL) while being treated with RBV/IFN or RBV/PEG-IFN for an anticipated period of 16 weeks were randomized to receive 40,000 titrated to 60,000 IU (if Hb did not increase by 1 g/dL after 4 weeks) by subcutaneous injection once a week EPO or matched placebo with titration for 8 weeks (double-blind phase DBP). Following the DBP, eligible patients received open-label EPO for the remainder of their HCV therapy. Patients randomized to receive EPO for 8 weeks could continue receiving EPO in the open-label period if they had a 1 point og greater increase in hemoglobon, showing that they were responding to the EPO. Patients randomized to placebo for 8 weeks could receive EPO in the open-label period if their hemoglobin was 12 g/dL or lower or if their RBV dose had been reduced. After HCV therapy cessation, patients were followed at 4, 12, and 24 weeks. 93 patients were randomized to EPO and 92 to placebo. So, the EPO treatment or the placebo was for 8 weeks.
Patients were excluded if they had a (1) contraindication for EPO including uncontrolled hypertyension. Rates of hypertension may be higher in HIV+ individuals; (2) iron deficiency and other treatable anemias; (3) significant atherosclerotic heart disease (contraindicating RBV use).
The primary endpoint of the study is the RBV dose maintenance at the end of the 8-week double-blind period. The secondary endpoint (at the end of DBP and 8-week open-label period) was any change in RBV dose from baseline, change in hemoglobin levels, and quality of life assessments (Linear Analog Scale Assessment [LASA] and Medical Outcomes Study Short Form-36v2, SF-36v2). This was an intent-to-treat analysis.
The definition of RBV dose success was patients with RBV dose equal to or greater than the dose at randomization. The definition of RBV dose failure was patients with RBV dose < the dose at randomization; all patients who withdrew before the end of week 4; all patients who withdrew due to anemia.
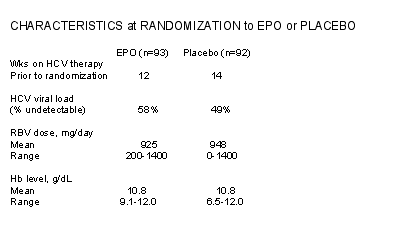
DEMOGRAPHICS of PATIENTS. The study did not include HIV/HCV coinfected patients. Mean age was 49-50 years (range 24-70). About 50% men. About 77% white, 11% Black, 10% Hispanic. Average weight 80 kg (1 kg=2.2 lbs). The patients in each group, placebo or EPO, had comparable baseline demographics.
34% patients in the EPO group and 37% in placebo had marked fibrosis or cirrhosis. Treatment naive: 69% in EPO, 60% in placebo. Treatment experienced: 31% in EPO group, 40% in placebo. Genotype 1: 71 in EPO and 76 in placebo. Genotype 2/3: 26 in EPO, 19 in placebo.
|
|
 |
| |
RESULTS
RBV DOSE SUCCESS. 88% (n=82) of patients who received EPO maintained (had RBV dose success) RBV dose at the end of the 8-week randomization period vs 60% of patients (n=55) who were randomized to placebo that were able to maintain RBV dose (p<0.001).
Mean RBV doses at the end of the DBP were 944 ± 217 mg/day (EPO) and 855 ± 241 mg/day (PL) (P<.001). At 8 wks, 80% of EPO-treated pts had a RBV dose ³ the dose at the start of HCV therapy vs 49% of pts on PL (P<.0001); mean changes from BL Hb were +2.2 ± 1.3 g/dL (EPO) and 0 ± 1.0 g/dL (PL).
Hemoglobin
Patients who received EPO saw their hemoglobin increase 2 points after 8 weeks and this was maintained through week 16. Patients who received EPO saw no improvement in hemoglobin until they received open-label EPO and then saw asimilar increase in hemoglobin. Mean Hb levels at the end of the DBP were 13.0 ± 1.3 g/dL (EPO) vs 10.8 ± 1.0 g/dL (PL) (P<.001).
--27% of patients receiving EPO saw 1 to <2 point increase in Hb vs 12% of patients who received placebo
--30% receiving EPO saw 2 to <3 point increase vs 3% receiving placebo
--27% receiving EPO saw 3 or greater increase in Hb vs 2% receiving placebo
--16% of patients receiving EPO saw <1 point increase in Hb vs 82% receiving placebo.
Quality of Life
At baseline patients had pretty severe quality of life, as expected. Mean scores on the LASA and selected SF-36v2 domains improved significantly from BL with EPO vs PL (P<.001-.003).
Using LASA assessment, patients receiving EPO had mean change from baseline in QOL scores of 12.7% vs 4.8% mean change in quality of life receiving placebo (P<0.001) improved quality of life. At week 17, both groups saw 17.8% mean change in quality of life. SF-36v2 evaluated at week 8 physical functioning, bodily pain, feeling of general health, vitality, social functioning, emotional well being, and mental health, and found significant benefits in these parameters for patients receiving EPO vs placebo. And after patients receiving placebo switched to EPO they showed mean improvements in quality of life scores.
Adverse Events
EPO appeared safe and well tolerated. There did not appear to be differences in adverse events between patients receiving placebo or EPO. More patients receiving placebo reported fatigue and anemia compared to patients receiving EPO. After switching from placebo to EPO fatigue improved. The study authors reported 1 serious adverse event that may be related to study drug: cerebrovascular disorder/cerebral thrombosis. There were several additional SAEs but they were deemed not related to study drug.
In summary the authors said: EPO resulted in (1) significantly greater proportions of patients maintaining or increasing RBV dose at the end of the 8 week double-blind period compared to their RBV dose at randomzation and the start of RBV/IFN therapy; (2) significantly greater increases in Hb at the end of the DBP; (30 similar results demonstrated in patients who crossed over from placebo to EPO; (4) and EPO was well tolerated.
|
|
|
|
|
|
|