 |
|
|
| |
Report from The 4th International Workshop on Clinical Pharmacology of HIV Therapy
Peter L. Anderson, Pharm.D., University of Colorado Health Sciences Center
|
| |
| |
The 4th International Workshop on Clinical Pharmacology of HIV Therapy was convened March 27th to 29th, 2003 in Cannes, France. The discipline of clinical pharmacology involves the study of how/why human patients respond to drugs including the study of how humans absorb, distribute, metabolize, and eliminate drugs (termed pharmacokinetics), and how the drug affects the patient (termed pharmacodynamics).
The meeting consisted of 4 plenary lectures, 26 oral abstracts, 44 poster abstracts, and several roundtable discussions led by panels of experts. Additional discussion and debate occurred during lengthy question-answer sessions after each oral abstract. Some of the abstracts contained science not directly applicable to clinical practice. These abstracts will not be covered in detail for this review, nor will studies with too few of subjects. The aim of this review is to summarize and briefly editorialize studies with more obvious clinical application and relevance for patients and doctors. The meeting organizers indicated that full access to all oral and poster abstracts will be made available at www.hivpharmacology.com (create a username and password; access is free). Bear in mind, much of the content at this meeting was hypothesis-generating, as opposed to large randomized prospective clinical trials. That said, many studies reported very intriguing data.
Note from Jules Levin: In this very interesting and well done 9,000 word report, Peter Anderson provides a comprehensive discussion of key studies presented at the workshop which have or may have clinical application now and in the future.
TOPICS:
-- Clinical pharmacology in developing countries
-- Pharmacogentics and drug transporters
-- HIV Drug Effectiveness and Genetic Differences: P-gp and MRP-2
-- Drug levels in women for nevirapine, efavirenz, and intracellularly for AZT & 3TC
-- Lopinavir and ritonavir levels (Kaletra) in women
-- Random levels of various antiretrovirals in peripartum females
-- Effect of weight on HIV drug levels
-- Hepatitis virus co-infection/liver function and drug levels: nevirapine, ritonavir, nelfinavir
-- TDM: therapeutic drug monitoring, including a table of therapeutic ranges of levels for protease inhibitors and NNRTIs
-- Drug levels and effectiveness for nevirapine and efavirenz
-- Indinavir drug levels evaluated in blood and hair
-- Inhibitory quotient (IQ) and effectiveness, which is the trough blood level/IC50 of the patient's virus, in boosted PI regimens including Kaletra, indinavir/r, saquinavir/r, and in non-boosted PI regimens
-- Drug levels and toxicity: Kaletra, Sustiva, Crixivan
-- Efavirenz drug levels when boosted by ritonavir
-- ddI/food/tenofovir interaction
-- Digoxin and ritonavir interaction
-- Indinavir/r and efavirenz interaction study
-- Liver transplants: tacrolimus, an immunosuppressive and HIV antiretroviral drugs
-- New antiretrovirals: pharmacokinetics (drug levels)
-- Ro334649 is a new saquinavir-analog protease inhibitor for protease inhibitor-resistant HIV; TMC114 is a new protease inhibitor that is also active versus protease inhibitor-resistant HIV; pharmacokinetics and safety of a new NNRTI, TMC125; he pharmacokinetics of the CCR5 antagonist UK-427,857 were described in healthy volunteers -- safety, food; the pharmacokinetics and drug-drug interaction profile of a new NNRTI, capravirine with Kaletra, nelfinavir;
--Atazanavir, a new PI about to be FDA approved, interactions with tenofovir, Invirase, and ritonavir
-- The new film-coated 625mg nelfinavir tablet: drug levels in blood and incidence of diarrhea compared to current nelfinavir formulation
-- Protein binding & Kaletra
-- A new analytical laboratory method to measure intracellular nucleoside-analog triphosphate concentrations
Clinical pharmacology in developing countries:
In the first plenary session, Terrence Blaschke from Stanford University described manufacturing issues for generic drugs in the developing world. Some generic companies can supply generic antiretroviral combinations for approximately $300 per year, compared with the ~$12,000/year cost in the US. Many of the companies that manufacture generic drugs are not under the auspices of an agency that assures product quality. This raises concern of inadequate manufacturing practices. To support this concern, Dr Blaschke quoted a WHO report that found no active drug in some antimicrobial and antimalarial generic stocks used in the developing world. The WHO has taken some initiative to investigate the content of generic products throughout the world, but more needs to be done. Additional information on this WHO initiative can be found at www.who.int/medicines/organization/qsm/orgqsm.shtml.
One abstract at this meeting described a quality-control analysis of generic nevirapine formulations obtained from the developing world. (1) Six stocks of nevirapine were collected from Kenya (Triomune 30ä, Cipla), South Africa (Viramuneä, Boehringer), and Zambia (Triomuneä, Cipla; Nevimuneä, Cipla; Nevirexä, Aurobindo Pharma Ltd). All formulations were labeled with 200 mg nevirapine. Some also had lamivudine and stavudine in the formulation, but data for these agents were not included in the abstract. Standard drug measuring techniques (HPLC) were used to quantify the amount of nevirapine in the formulations. The average (range) nevirapine content of the formulations was 197.9 mg (194.2 to 205.5 mg). All formulations were in the expected and desirable ranges.
In a second plenary talk, Ceppie Merry from St James's Hospital in Dublin, Ireland described her recent efforts to establish a program by which antiretroviral agents could be more widely utilized in Africa. One effort is to provide a central AIDS treatment information center for Africa that local clinicians can contact for information on what drugs are available in their area and in what quantities, and also for traditional drug information. Another investigator from Zimbabwe presented two abstracts that described initiatives to improve adherence in his homeland. One involved training volunteer lay people in a Zimbabwe community to promote adherence in HIV infected patients. (2) The other described an effort to form a more structured adherence clinic, based on a model at the University of Buffalo, NY where he was trained. (3)
Pharmacogentics and drug transporters
Two plenary presentations were devoted to pharmacogenetics. The aim of pharmacogenetics is to discover differences among people at the genetic level that causes differences in drug response. For example, enthusiasm currently exists for the possibility that a certain HLA genotype pattern may one day predict who will experience the abacavir hypersensitivity reaction. (4) Richard Kim from Vanderbilt University spoke on pharmacogenetics of drug transporters. P-glycoprotein (P-gp), for instance, is a drug transporter that likely evolved to protect humans by countering the absorption of ingested plant or microbe poisons and by limiting penetration to the brain, gonads, and developing fetuses. The pump also clears poisons into urine or bile. It turns out that many therapeutic drugs, including HIV protease inhibitors are actually good substrates for P-gp (5). A P-gp substrate means that P-gp recognizes the drug and performs its pumping activity on the drug. PIs are P-gp substrates. Digoxin is a P-gp substrate. So, the activity of protease inhibitors could be compromised in patients that have high expression/function of P-gp because this drug transporter could pump protease inhibitors out of places like cells where you would want them. Kim spent significant time describing his work, which currently involves the biochemical mechanism for how inducing drugs (e.g., rifampin) increase the expression of drug metabolizing enzymes and drug transporter proteins (including P-gp). It is certainly a more complicated system than previously appreciated. First, there are dozens of different drug transporters just as there are dozens of different drug metabolizing enzymes. Second, the biochemical mechanism that determines the amount of drug metabolizing or transporting enzymes a patient will express involves multiple interplaying nuclear co-factor proteins that initiate gene transcription. Lastly, people can vary genetically throughout this whole system. (6-8)
The other plenary presentation was given by Amalio Telenti from Switzerland. He also addressed drug transporter systems in humans, but approached the topic as a clinician. Telenti described his published results of a large epidemiological survey of HIV-infected patients treated with efavirenz- or nelfinavir-based therapy that found an association between a P-gp gene (MDR-1) mutation, TT at nucleotide position 3435, and a significantly better CD4 recovery than people with CC at nucleotide position 3435. (9) This seemed biologically plausible for nelfinavir because the TT at 3435 had been associated with lower P-glycoprotein expression in other studies, which would allow more nelfinavir to be absorbed from the gut and distributed throughout the body. (10) This would presumably enhance drug response. However, Telenti also measured drug concentrations in their study and paradoxically found significantly lower nelfinavir concentrations in the 3435 TT group. It was also paradoxical that efavirenz was related with 3435 at all because it is not a substrate of P-gp. He discussed that other studies have also reported conflicting results regarding 3435 TT / CC and P-gp substrate blood levels. He stated that future studies will need to consider ethnicity, multiple genes, and multiple genetic mutations. The main point from both plenary lectures was that the scientific framework for association studies between genetics of drug metabolizing or transporter enzymes and drug response is only in early development. Much more work is needed before clinical implications can be deciphered.
HIV Drug Effectiveness and Genetic Differences: P-gp and MRP-2
Several abstracts at this meeting described an association between genetic variability in the P-gp gene, MDR-1 and protease inhibitor blood concentrations. Focus is shifting from the C3435T nucleotide position, which does not encode a change in the protein structure, to a linked genotype, G2677T (in this case, linked indicates that a DNA strand with C at 3435 will most likely carry a G at 2677, whereas a strand with T at 3435 will most often carry a T at 2677). The 2677 G®T mutation encodes a change in the protein structure of P-gp. (6) In one abstract describing 33 antiretroviral naïve subjects in a study of zidovudine, lamivudine, and indinavir therapy, significantly lower blood clearance of indinavir (higher blood concentrations) was found in four patients with TT at 2677 compared with 12 patients with GG (p=0.02); no effects were found for the 17 patients with GT. (4) This study also found a larger reduction in HIV-RNA from baseline to one-year in the TT and GT patients compared with the GG patients (P<0.05). (11) Another investigator studied 34 lopinavir/ritonavir/saquinavir-treated patients and the influence of the G2677T genotype on protease inhibitor blood levels. The same trend was found; higher drug levels in persons with 2677 TT versus GG, but the differences were not statistically significant. These investigators also studied the effects of genetic variability in the gene encoding another drug transporter, MRP-2 on protease inhibitor drug levels. MRP-2, a transporter with functional similarities with P-gp, is emerging as another important protease inhibitor transporter. (12) They found 3-fold higher saquinavir trough concentrations in persons with GG at position 1249 of the gene compared to persons with GA or AA (P=0.009). (13) This is the first description of such a relationship between genetic variability in the MRP-2 gene and antiretroviral drug concentrations, to my knowledge.
Another abstract reported very interesting results regarding MRP-2. It is believed that the MRP-2 transporter has two active sites, one that pumps the drug across the membrane, and another site that greatly stimulates that pumping action. These authors hypothesized that probenecid binds to this second site and greatly stimulates the activity of the pump. Ritonavir, saquinavir, plus or minus probenecid were given to mice. When probenecid was present, saquinavir concentrations were reduced by > 10-fold. (14) This is a dramatic reduction in plasma exposure. Meeting participants were concerned because probenecid is a drug that could possibly be given concomitantly with protease inhibitors in clinical practice. The investigator suggested that the doses of probenecid and protease inhibitors could be separated to lessen the magnitude of a possible interaction. Information about this interaction in humans is needed. In speaking with the study author directly he said they think that in their
rat experiments the uptake of ritonavir from the gut is limited by probenecid
and therefore ritonavir levels may drop. This may limit ritonavir's boosting
capacity.
Two abstracts investigated the association between P-gp expression and HIV-infection. One abstract measured MDR-1 mRNA expression in PBMCs of healthy volunteers versus HIV-infected patients and found evidence of decreased expression (~2-fold) in HIV-infected patients. (13) However, another paper measured MDR-1 mRNA in placenta tissue of healthy controls versus HIV-infected women. (15) They reported HIV-infected women had significantly higher MDR-1 mRNA levels (~20-fold). Evidently, more work is needed to clarify the biological effects of HIV-infection on MDR-1 expression throughout multiple human tissues.
Another study tested whether exposing PBMCs to fairly high levels of protease inhibitors changed the expression of P-gp protein on the cell surface (ex vivo). Of all marketed protease inhibitors, only nelfinavir altered P-gp expression with statistical significance; expression was increased. (16) The C3435T genotype was determined in 12 of 15 healthy volunteers who donated the PBMCs for the study. Geneotypes at this position did not alter the results. The clinical significance of these data are yet unknown.
Pharmacokinetics in Special Populations:
Several intriguing studies were reported in this session. One theme was sex differences in drug levels that correspond with, and provide hypothetical explanations for, the exaggerated drug responses observed in females compared with males. Another important theme was differences in drug levels according to the presence of hepatitis virus infection.
Sex and drug levels:
An oral abstract described significantly higher random nevirapine blood levels among 100 female (median 6.7 mcg/mL) compared with 268 male (5.5 mcg/mL) patients in the Netherlands. (17) The authors set a nevirapine threshold concentration of > 6.0 mcg/mL to indicate a "toxic" level (therapeutic drug monitoring and therapeutic ranges are commonplace in many European countries). Using logistic regression for "toxic" level versus sex, age, and weight, only sex was related with "toxic" drug levels (P=0.02). These results were essentially duplicated for patients taking efavirenz. (18) The median level from 38 females was 3.0 mcg/mL compared with 2.3 mcg/mL among 156 males. A "toxic" level for efavirenz was set at 4.0 mcg/mL. Using logistic regression, female sex was independently associated with a "toxic" efavirenz level (P=0.03). It was pointed out that epidemiological studies suggest that females have more severe rash and liver toxicity during nevirapine therapy. The findings in this study might shed light on an explanation for these observations. It was unclear why females had higher blood levels. One possibility is that they adhere to therapy better than males. Overall, this study raises important questions, however, the data were generated from retrospective analyses and must be confirmed with controlled studies.
Another study reported a sex difference in intracellular zidovudine- and lamivudine-triphosphate concentrations among patients in a study of zidovudine, lamivudine, and indinavir therapy. (19) Nucleoside analogs must be phosphorylated in the cell to the active triphosphate moiety, which exerts both therapeutic and toxic effects. At planned intervals, this study collected PBMCs for zidovudine- and lamivudine-triphosphate measurement in 33 subjects. A total of 310 lamivudine-triphosphate and 282 zidovudine-triphosphate concentrations were obtained (average of ~9 each/subject). The half-lives were estimated at 7 hours for zidovudine-triphosphate and 22 hours for lamivudine-triphosphate. Females (n=4) had about 2.3-fold higher zidovudine-triphosphate (P=0.003), and 1.6-fold higher lamivudine-triphosphate (P=0.002) compared with males (n=29). There were no sex differences in demographics or the plasma concentrations of zidovudine, lamivudine, and indinavir. Females reached < 50 of HIV-RNA in half the number days as compared with males (p=0.02), which remained significant when adjusted for baseline HIV-RNA. It was pointed out that epidemiological studies find females experience more exaggerated virologic responses and toxicity to nucleoside analogs; including nucleoside analogs used for cancer. A controlled study is needed to confirm these findings.
Conversely, a small study did not detect a sex difference in lopinavir concentrations in 30 patients receiving a lopinavir-based regimen. (20) Samples were collected 10 to 14 hours after lopinavir dosing. Although females had higher lopinavir concentrations than males (6.4 versus 5.9 mcg/mL), but the difference was not significant. No effects of weight or age were found with regard to lopinavir concentrations, whereas an inverse linear relationship was found between weight and ritonavir concentration (P<0.05). Others have reported a major weight effect on lopinavir plasma levels; this study may have been under-powered to detect lopinavir differences.
Another study collected random levels of various antiretrovirals in peripartum females (n=103). (21) Levels in the mother, in cord blood, and in amniotic fluid were obtained. Cord blood/maternal ratios were high for zidovudine (1.22), stavudine (1.32), lamivudine (0.93), and nevirapine (0.88), whereas ratios were low for didanosine (0.38), nelfinavir (0.24), and amprenavir (0.27). Lamivudine concentrations were approximately 4-fold higher in amniotic fluid compared with cord blood and maternal blood. The unplanned, observational nature of the study and the mixture of various drug combinations make it difficult to draw solid conclusions from this study, but these results are similar to findings in past studies.
Ethnicity and drug levels
A study from Israel described differences in nelfinavir and lopinavir blood levels in Caucasian (n=51) versus Ethiopian (n=57) origin patients. (22) Ethiopian persons had higher trough and peak concentrations of both nelfinavir (P<0.09) and lopinavir (P<0.004) compared with Caucasians (11.2/12.4 and 1.6/2.8 compared with 5.5/7.9 and 0.9/1.8 mcg/mL, respectively). Differences in weight between these ethnicities would explain some of these results and differences in diet could have also played a role. However, these results warrant controlled studies to investigate ethnicity differences in the pharmacokinetics of antiretrovirals.
Hepatitis virus co-infection/liver function and drug levels
Several studies investigated pharmacokinetics of antiretrovirals in patients with hepatitis virus infection or with elevations in liver function tests. One study investigated nelfinavir blood levels in 50 HIV/HCV-, 29 HIV/HCV+ (no cirrhosis), and 14 HIV/HCV+ (biopsy-confirmed cirrhosis) infected patients. (23) Serial nelfinavir and M8 active metabolite levels were obtained after dosing. Nelfinavir and M8 were summed and this AUC was found to be lowest in the HIV/HCV- group, 1.5-fold higher in the HIV/HCV+ (non-cirrhosis) group, and 2.5-fold higher in the HIV/HCV+ (cirrhosis) group (P<0.05). Weight was accounted for in the oral clearance calculations. Most of the difference among the groups was due to changes in nelfinavir concentrations (not as much from M8). The investigators suggested that nelfinavir doses may need to be decreased in HIV-HCV+ patients, but several participants questioned that conclusion because nelfinavir is not toxic at higher doses and it has potency problems at usual doses.
Abbott laboratories studied ritonavir pharmacokinetics in 6 HIV-infected subjects with normal lever function and 6 HIV-infected subjects with mild hepatic impairment (Child-Pugh 5-6). (24) Two impaired subjects had HBV and four had HCV-infection; one control subject had HCV-infection. Ritonavir pharmacokinetics were tested after a single dose and again at steady-state (after 2 weeks). After a single dose, the AUC was significantly higher in the subjects with hepatic impairment compared with controls (126.5 versus 99.8; P=0.03). However, there were no differences between the groups at steady-state. Plasma protein binding was not statistically different between the groups. The investigators then conducted a second study in HIV negative subjects, 6 controls and 6 with moderate hepatic impairment (Child-Pugh 7-9; 3 had HCV-infection). Curiously, ritonavir concentrations were lower in the moderate hepatic impaired group (AUC 41 versus 66; P=0.08). Protein binding and trough concentrations were not different. Although the authors suggest that ritonavir dose adjustments are not needed for mild or moderate hepatic impairment, more studies are clearly needed to characterize the pharmacokinetics of protease inhibitors in patients with liver dysfunction.
Another study investigated relationships between nevirapine total and plasma unbound trough concentrations and liver function tests in 82 HIV-infected subjects (41% HCV or HBV+). (25) Higher nevirapine trough concentrations were observed in patients with HBV or HCV infection (P=0.025). Changes from baseline in Gamma-GT but not ALT were related with nevirapine concentrations. This may be because nevirapine is a hepatic inducer. These data provide more motivation to study antiretroviral pharmacokinetics in patients with liver disease/dysfunction.
Pharmacokinetics and toxicity/efficacy and therapeutic drug monitoring (TDM)
Many studies at this meeting addressed relationships between antiretroviral blood concentrations and toxicity/efficacy and also the strategy of TDM. These topics are closely related because concentrations that determine efficacy and toxicity must be defined in order to implement TDM. The aim of TDM is to individualize patient doses, based on the person's capacity to absorb, metabolize, and excrete the drug, to maintain "therapeutic" drug levels. TDM is commonplace in several Western European countries, but is not particularly common in the US.
Drug levels and efficacy: NNRTIs
Random nevirapine and efavirenz concentrations were obtained on three occasions in a large heterogeneous French cohort. (26) There were 227 persons who received efavirenz, 186 were NNRTI-naïve, whereas 130 persons took nevirapine, 120 of these were NNRTI-naïve. The authors set the "therapeutic" blood concentrations for these agents at 1.1 mcg/mL for efavirenz and 4.0 mcg/mL for nevirapine. 90% of efavirenz patients and 54% of nevirapine patients had "therapeutic" blood levels (P<0.001 for the difference). In a logistic regression among the NNRTI-naïve subjects for having < 200 HIV-RNA by week 12, prescription to efavirenz was significantly related with success (OR=4.37; 95% CI=2.76 to 6.9). The authors concluded that the higher incidence of "sub-therapeutic" nevirapine blood levels might explain these virologic results. The audience suggested the nevirapine "therapeutic" target was set too high, because many investigators use 3.4 mcg/mL versus 4.0. The presenter indicated he had tested the results with 3.4 mcg/mL and this had not changed the outcome. Another observational study measured 447 NNRTI plasma levels (328 efavirenz, 119 nevirapine) in a heterogeneous cohort of patients. (27) The therapeutic ranges set by the presenter were 1.1 to 5 mcg/mL for efavirenz and 3 to 8 mcg/mL for nevirapine. 30% of the NNRTI levels were below these thresholds. In a sub-analysis of just the NNRTI-naïve patients, those with undetectable HIV-RNA or the "largest HIV-RNA decrease" had higher efavirenz levels compared with their counterparts (1.77 versus 1.49 mcg/mL; P<0.05).
Drug levels and efficacy: Protease inhibitors
Another study investigated indinavir concentrations in the hair of a heterogeneous group of patients and correlated these levels with therapeutic outcome. (28) Patients were treated with indinavir/ritonavir 200-800mg/100mg. Indinavir hair concentrations (in mcg/g) were related with indinavir plasma trough (P=0.03), the cumulative indinavir dose (P=0.004), the duration of indinavir treatment (P<0.01) and the viral load (P=0.02). Another group of French investigators investigated the 4-month virologic response according to indinavir blood levels, self-reported adherence, and baseline demographic/disease characteristics in protease inhibitor-naive patients initiating indinavir-based therapy. (29) In multivariate analyses, < 500 HIV-RNA at 4 months was only related with "100%" adherence level (based on patient questionnaire): OR 8.8; P=0.0002. When looking at 100% adherent patients only, then virologic success was related with duration of baseline NRTIs, baseline HIV-RNA, and the ratio of expected indinavir concentration to observed concentration; for example, the threshold indinavir trough in this analysis was 0.184 mcg/mL. It is important to bear in mind that none of these were prospective controlled studies.
The inhibitory quotient (IQ) and efficacy
A current concept for therapeutic drug levels in experienced patients is that the susceptibility of the virus must be considered. This spawned the inhibitory quotient (IQ), which is the trough blood level/IC50 of the patient's virus. Presumably, if the patient's HIV IC50 is increased, the trough concentration could be increased to maintain antiviral activity. Several retrospective studies found a superior association between the IQ and virologic response compared with drug levels alone or viral phenotype alone and virologic response. One paper described standardized IQ values amongst several ritonavir-boosted protease inhibitor regimens. (30) It is critical to use the same methods to calculate IQs including, the cell lines used for the phenotype assay; the concentration of plasma protein-binding proteins in the phenotype assay; and how the blood levels are measured and calculated. The authors made efforts to standardize all these variables and found that lopinavir/r 400/100 twice daily had the highest IQ for wild-type virus (HIV pNL4-3); for example lopinavir IQ was 67.4, indinavir/r 800/100 was 13.9; saquinavir/r 1600/100 qd was 1.3. For unboosted protease inhibitors, atazanavir was highest, for example, atazanavir was 10.0, nelfinavir 1.0, and the new fosamprenavir 0.7. Prospective studies are needed to determine the clinical significance of IQs.
Drug levels and toxicity
Two studies investigated a relationship between lopinavir blood levels and lipid elevations. In a prospective observational study, 30 patients were followed for 12 months. (31) Lipids (TG, HDL, LDL, & total) and lopinavir trough blood levels were measured at baseline and months 0, 1, 3, 6, and 12. No correlations were observed between lopinavir trough blood levels and changes in lipid parameters. Another retrospective study investigated the changes in HDL, TC/HDL, and TG (triglycerides) in 142 heterogeneous lopinavir-treated patients. (32) Overall by month 12, they detected a mean 21% increase in HDL (good cholesterol) per patient; the ratio TC/HDL was also increased from 5.53 at baseline to 6.11 at month 12 (P=0.03); and TG increased from 3.02 to 6.78 mmol/L (P=0.04). In 30 patients lopinavir concentration data were available at month 3, and in 15 patients data were available at month 6. Persons with TG³2.3mmol/L at these time points had higher lopinavir trough concentrations compared with patients with TG<2.3 (6.78 versus 3.02 mcg/mL at month 3; P=0.05 and 9.19 versus 0.96 mcg/mL at month 6; P=0.02). The value of 0.96 mcg/mL for lopinavir is highly indicative of non-adherence. These results should be confirmed with prospective controlled studies.
A number of studies investigated drug levels in blood for relationships with other toxicities. A prospective observational study investigated the effects of efavirenz blood levels on neuropsycological tests in 23 consecutive patients starting efavirenz. (33) Changes in test scores from baseline to weeks 2, 4, and 12 were calculated. Efavirenz blood levels were obtained approximately 12 hours post-dose at weeks 2 and 4. No relationship between changes in test scores and efavirenz blood levels could be identified. Another study investigated lopinavir, ritonavir and efavirenz blood levels and a lipodystrophy score in 30 patients. (34) Unfortunately, nucleoside analogs were not studied. These subjects were highly heterogeneous in terms of disease severity, duration of past therapy, duration of HIV disease, and the current regimens. No relationships between blood levels of these drugs and lipodystrophy score were identified. Another study described a higher incidence of nephrolithiasis in 12 subjects treated with indinavir/ritonavir (doses not provided) compared with 37 subjects on standard IDV alone (RR=3.66; 95% CI=0.96 to 14.05). (35)
TDM
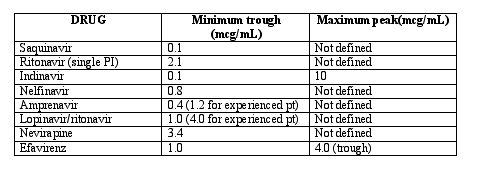
There was some expert discussion on therapeutic ranges and TDM. First, most experts suggested TDM only for special scenarios; for suspected drug-drug interactions, in patients with severe hepatic or renal disease, in pediatrics, or in pregnancy. Care must be taken to record the exact time of last dosing and the time of blood collection. A therapeutic range should be identified (see table), and it traditionally includes a certain trough threshold for viral efficacy and peak threshold for toxicity. Peak levels generally occur ~1 hour post-dose when drugs are administered in the fasted state, and ~2 to 6 hours post dose when administered with a meal. The following table contains suggested therapeutic ranges for the naïve patient (wild-type HIV). It must be acknowledged that a drug level in this range does not guarantee the desired response. More information about this table is available at www.hivpharmacology.com (create a username and password; access is free).
|
|
 |
| |
Drug interactions: NNRTIs
One study retrospectively investigated efavirenz trough concentrations in 48 patients taking or not taking boosting doses of ritonavir and undergoing routine TDM monitoring. (36) The efavirenz Cmin was significantly higher in the ritonavir boosted group versus the group not on ritonavir (3.15 versus 2.22 mcg/mL; P<0.05). Side effects were not different between the groups.
Drug interactions: ddI/food/tenofovir
Bristol Myers Squibb reported results on a randomized open-label cross over drug interaction study with enteric-coated ddI and tenofovir in 36 healthy volunteers. (37) All drug-drug interaction data were compared with the reference ddI EC on an empty stomach. The tenofovir and ddI EC doses were administered with a small meal (373 kcal). Compared with ddI EC while fasted, all the tenofovir and ddI EC plus food studies showed a lowering of the Cmax and an elongation of the plasma ddI half-life (~40%). Therefore, the plasma ddI AUC was approximately equivalent for ddI EC 400mg fasted and ddI EC 250mg plus tenofovir plus food (AUC ratio 0.953; 90% CI 0.87 to 1.04). The ddI EC 325mg plus tenofovir plus food gave a higher plasma ddI AUC compared with ddI EC 400mg fasted (AUC ratio 1.13; 90% CI 1.03 to 1.24). Relative to ddI EC 250mg fasted, ddI EC 200mg plus tenofovir plus food gave a higher AUC (AUC ratio 1.16; 90% CI 1.06 to 1.27). Therefore, based on AUC ratio, 250mg ddI EC plus tenofovir plus food is equivalent to 400mg ddI EC while fasting. For patients who need 250mg ddI EC, a dose reduction to 200mg ddI EC plus tenofovir plus food results in a slightly increased ddI AUC. Another study measured random ddI (n=62) and tenofovir (n=57) plasma concentrations in 54 HIV-infected patients treated with 400mg ddI EC and standard tenofovir. (38) The two drugs were taken separately, ddI EC while fasting and tenofovir with food. Compared with median "historical" ddI data, the median ddI concentrations in this study were 2.6-fold higher (P=0.003). Tenofovir concentration data were not different between historical controls and the results in this study. It should be noted that a planned prospective study would be needed to accurately quantify this interaction in HIV-infected patients. It must also be stressed that no intracellular triphosphate studies of the interaction between ddI and tenofovir in patients have been conducted. These agents are both adenosine analogs a may compete with some of the same enzymes for intracellular phosphorylation.
The effect of a qd regimen of indinavir/ritonavir (1200/400mg) with food on ddI plasma levels in 8 healthy volunteers taking ddI EC was determined in a cross-over study. (39) Combined doses of all drugs were either taken together with food, or taken 2 hours after a meal. The ddI plasma levels were compared with ddI EC during fasting. The combination of ddI EC and indinavir/ritonavir with food was associated with ddI plasma levels that were nearly identical with ddI EC taken alone during fasting.
Drug interactions: Protease inhibitors
A drug interaction study between ritonavir and digoxin was reported. (40) Ten healthy volunteers participated. At steady-state, digoxin AUC was increased by approximately 23% when dosed with ritonavir (200mg) compared with digoxin alone (P=0.04). In the clinic setting, patients treated with digoxin should have digoxin blood levels followed if ritonavir or other protease inhibitors are initiated or discontinued.
Some descriptive protease inhibitor associated drug-drug interaction studies were presented. In 11 highly experienced HIV-infected patients, the pharmacokinetics of lopinavir/r 400/100mg and indinavir 600 mg bid were characterized. (41) The drugs were given with a light breakfast. Compared with healthy volunteer studies, the concentrations in this cohort were lower (up to 64% for some parameters). The average trough concentrations for indinavir and lopinavir were 0.3 and 3.1 mcg/mL, respectively. A similar study used the same drugs except indinavir dose of 800mg in HIV-infected patients. (42) The indinavir trough was 0.61 (range 0.05 to 1.91) mcg/mL and the lopinavir trough was 2.9 (0.55 to 4.4) mcg/mL. Again, these concentrations were slightly lower than expected. One other study described pharmacokinetics of indinavir (400mg bid) and lopinavir/ritonavir (400/100mg bid) in 10 HIV-infected patients. (43) Lopinavir concentrations were obtained before and after indinavir was added. In contrast to the other studies, the concentrations of indinavir and lopinavir were very close to what would be expected based on past studies. The median trough of lopinavir was 5.6 and 7.3 mcg/mL before and after indinavir and the median trough of indinavir was 0.45 mcg/mL. This study also measured indinavir and lopinavir levels in CSF and semen. Lopinavir concentrations were mostly undetectable in CSF and were about 0.25 mcg/mL in semen. As expected, indinavir penetrated into semen and CSF (medians of 0.59 and 0.04 mcg/mL, respectively).
In another study, indinavir pharmacokinetics and efavirenz levels were described in 6 HIV-infected patients treated with indinavir/ritonavir and efavirenz (plus or minus d4T). (44) Indinavir/ritonavir (800/100mg) was given with food. The mean indinavir trough was 0.32 (range 0.07 to 0.98) mcg/mL. This value is lower than that reported for these doses without efavirenz (~0.9 mcg/mL), and one person had a level below the supposed therapeutic cut-off for naïve patients of indinavir (~0.15 mcg/mL). This indicates that in the presence of efavirenz, higher doses of ritonavir may be needed to offset the inducing properties of efavirenz on indinavir. Efavirenz concentrations were in the expected range (1.1 to 7.9 mcg/mL). Another study described saquinavir concentrations in 8 HIV-infected patients taking saquainvir (Invirase) 600mg with lopinavir/ritonavir 400/100mg bid. (45) Troughs (10 to 14 hours psot-dose) levels were obtained and compared with previous data with saquinavir/ritonavir 600/100mg bid. It was found that ritonavir concentrations were decreased (no statistics), but that saquinavir and lopinavir were comparable with historical controls.
An interesting study reported on seven HIV-infected patients who had had a liver transplant and were taking tacrolimus, an immunosuppressive with their antiretroviral regimen. (46) Tacrolimus dosing and blood level data were compared with those from a group of HIV negative controls. The antiretroviral regimens varied. The doses of tacrolimus needed to achieve target blood levels were very different according to concomitant antiretroviral drug. Compared with the tacrolimus dose in non HIV-infected patients, a dose of 13% that value was used with nelfinavir or amprenavir; 4% that value for lopinavir/r; 54% that value for trizivir; 200% that value for efavirenz; and about the same dose for nevirapine. These data are consistent with the known drug-metabolism inhibiting potential of the protease inhibitors and the inducing potential of efavirenz.
New antiretrovirals: pharmacokinetics
The pharmacokinetics of several new agents or formulations were presented. Ro334649 is a new saquinavir-analog protease inhibitor for protease inhibitor-resistant HIV. (47) In healthy volunteers, it was found that this agent is well-absorbed with or without food, although the absorption variability was improved with food. Ritonavir boosted Ro334649 exposure 4.5-fold. Safety was not reported. This compound is in clinical development. TMC114 is a new protease inhibitor that is also active versus protease inhibitor-resistant HIV. (48) In 76 healthy volunteers, it was found that TMC114 induces its own metabolism. It was also apparent that TMC will likely require boosting with ritonavir, which increases its exposure by at least 3-fold; for example, 1200mg TMC114 tid alone gave trough concentrations of 0.142 mcg/mL, whereas 1200mg/200mg TMC114/ritonavir qd gave a trough of 0.358 mcg/mL. Adverse effects were decreased when co-dosed with ritonavir because of PEG4000 in liquid TMC114 solution. Presumably, the ritonavir combinations consisted of smaller doses of PEG4000, which accounted for the improved tolerability. TMC114 was recently reformulated into a solid tablet. TMC114 side effects were typical for protease inhibitors, gastrointestinal, perioral paresthesia, increased lipids. Rash was also observed. The pharmacokinetics and safety of a new NNRTI, TMC125 as mono-therapy in 12 HIV infected antiretroviral naïve subjects was described. (49) Dosing was 900mg bid. Plasma levels were obtained at 7 days. The half-life was estimated at 30 to 40 hours. The average drop in HIV-RNA at 7 days was 2.0 logs. No relationship could be identified between the trough concentrations of TMC125 and the drop in HIV-RNA. Side-effects were gastrointestinal (also presumed from PEG4000 co-formulation), somnolence, and pruritis. A new tablet formulation has been developed.
The pharmacokinetics of the CCR5 antagonist UK-427,857 were described in healthy volunteers. (50) The drug was rapidly absorbed (peak occurs at 0.5 to 4 hours) and its half-life was 19 hours. Food significantly reduced the rate and extent of absorption. The highest doses caused postural hypotension, asthenia, dizziness, abnormal vision, and headache reported. No laboratory abnormalities were found including no "clinically relevant" changes in QTc interval. The conclusion was that these results warranted further development.
Two posters described the pharmacokinetics and drug-drug interaction profile of a new NNRTI, capravirine. Capravirine has activity versus viral mutants including the K103N. One poster investigated the combination of nelfinavir plus capravirine in healthy volunteers. Descriptive data in HIV-infected patients were also provided. (51) Capravirine AUC concentrations were increased by about 2.4-fold with nelfinavir. Nelfinavir concentrations were unchanged, however, the active metabolite M8 concentrations were decreased by about 25% during capravirine treatment. In HIV-infected patients treated with identical dosing, capravirine concentrations were slightly lower than in the healthy volunteer counterparts (AUC 21.8 versus 32.7; no statistics reported). The mechanism for this difference was not known. Another study investigated a complex drug-drug interaction between lopinavir/ritonavir and capravirine. (52) Data were gathered from several studies in healthy volunteers and HIV-infected subjects. A math model was constructed to describe the various drug interactions. The bottom line was that capravirine induces the metabolism of both lopinavir and ritonavir. It appears that a dose increase to Kaletra to 533/133 bid will compensate this interaction. Going the other way, ritonavir increases capravirine concentrations, but not as much when combined with lopinavir. Current dose-ranging phase II studies are underway to identify a capravirine dose when combined with Kaletra.
Two posters described pharmacokinetic and drug interaction characteristics of atazanavir. One study measured the effect of tenofovir on atazanavir and ritonavir pharmacokinetics in heavily pre-treated patients taking atazanavir (300mg qd), ritonavir (100mg qd), and various NRTIs. (53) The atazanavir AUC was reduced by 25% after tenofovir was added. The levels of ritoanvir were also lower, but not with statistical significance. Another poster described saquinavir plasma levels in 11 patients taking the Invirase formulation with atazanavir. (54) Several doses were studied: 1200 and 1600mg qd Invirase or 600 mg tid Inverase plus 400mg ataznavir qd all with food. Some levels were obtained after observed dosing of 1200mg Invirase plus 400mg atazanavir qd. A 3 - hour post-dose concentration was obtained (median 630 mcg/mL) and the 24-hour concentration was 0.06 mcg/mL. These data provide evidence that atazanavir increases Invirase concentrations, however, more studies will be needed to determine appropriate doses and dosing frequency.
Roche pharmaceuticals presented information on a new film-coated 625mg nelfinavir tablet. In 52 healthy subjects, the AUC and Cmax of nelfinavir were equivalent between the 250mg Viracept formulation and the new 625mg film-coated tablet when dosed with a 800kcal meal (Cmax and AUC ratios, 101% and 95%, respectively). (55) The active M8 metabolite concentrations were slightly lower with the 650 mg tablet compared with the 250 mg formulation (AUC 6.4 versus 7.6), but no statistics for the comparison were reported. Interestingly, there was evidence that the 625 mg film-coated tablet might cause less diarrhea. A two-arm observation prospective study of gastrointestinal tolerability of the new 625 mg formulation was conducted in HIV-infected patients. (56) In one arm, patients who switched from the 250mg tablets to the new 625 mg tablets were followed for gastrointestinal tolerability using a stool diary. Another arm followed stool diaries in a group of patients who initiated nelfinavir as the 625mg tablet. After the switch to the new formulation in the first arm, the percentage of patients without diarrhea increased from 48% to 74% (statistics not provided). In the other arm, approximately 75% of patients had no diarrhea before commencing nelfinavir, and approximately 50% of patients had no diarrhea in the first two weeks after initiating 625 mg film-coated nelfinavir therapy (statistics not provided). The presumed mechanism for the improved tolerability owed to differences in excipients (pill fillers/additives) between the formulations. Roche states the new formulation will be filed for approval in Europe this May. No information was given about its prospects with Pfizer in the US.
Miscellaneous
The plasma protein binding of protease inhibitors influences the amount of drug that can leave the blood and enter cells and HIV infected tissues. One study at this meeting found that lopinavir is more highly bound to plasma proteins at the trough concentration compared with at the peak concentrations in HIV infected subjects. (57) Presumably, when concentrations become very high, the plasma protein sites become saturated and a higher fraction of drug is in the unbound state. The clinical significance of this finding is not known. Another study investigated the effects of differing AAG concentrations in patients (this is the main plasma protein that protease inhibitors bind to) and the effects on nelfinavir protein-binding. No relationships were identified. (58) Protein-binding is definitely an important drug characteristic, but the few studies that have investigated differences in protein-binding among patients have not yet established its clinical relevance.
One of the most intriguing abstracts at this meeting, in the opinion of this attendee, involved findings from recent studies of intracellular nucleoside-analog triphosphate concentrations in the PBMCs (a type of cells) of HIV-infected patients. These studies are analytically challenging, and therefore few data of this kind exist. A laboratory in France has developed a new analytical method to measure intracellular nucleoside-analog triphosphate concentrations. In an abstract, they reported that patients taking zidovudine therapy were chemically converting about 30% of the zidovudine-phosphates in the cell to stavudine-triphosphate. (59) This is an amazing finding that should be confirmed by other laboratories. The authors also reported a half-life for intracellular d4T-triphophosphate of approximately 7 hours, and over 24 hours for ddA-triphosphate (active anabolite of ddI). These are the first data of this kind in patients. When one considers the amazing findings in this study along side the findings described earlier of significantly higher intracellular zidovudine and lamivudine-triphosphates in females versus males, it very motivating to conduct more research in this area. It should be remembered that nucleoside analogs continue to comprise the backbone of nearly all our HIV regimens. Indeed, most regimens contain two active nucleoside analogs and only one other active drug.
Meeting Summary:
There were several key findings reported at this meeting in the view of this attendee. Preliminary evidence continues to mount that protease inhibitor blood levels, and by extension antiviral activity, are determined in part by the expression and genetic variability in the drug efflux pumps P-gp and more recently MRP-2. Expect much more work involving P-gp and MRP-2 in the future including, drug-drug interaction studies at the level of MRP-2. Another key finding was of sex differences in NNRTI blood levels and intracellular nucleoside analog triphosphate concentrations. In both cases, females had higher levels than males, and the findings were consistent with epidemiological reports of enhanced therapeutic/toxic responses in females compared with males. Along similar lines, several studies also found higher antiretroviral blood levels in patients co-infected with hepatitis C or B. These are areas where more data are urgently needed, because of important therapeutic implications (e.g., are dose adjustments needed for hepatitis-C co-infected patients). This meeting also reminded attendees of the importance of studying intracellular nucleoside analog triphosphate concentrations. Besides the above-mentioned sex difference in triphosphates, another investigator described d4T-triphosphate concentrations in patients not taking d4T but instead taking zidovudine (d4T-triphosphate concentrations were 30% of the zidovudine-triphosphate levels). Much more research activity is needed in this area. Finally, the reports on drug levels and efficacy were dominated by NNRTIs, an apparent shift from previous meetings that focused on protease inhibitors. It is possible that NNRTIs may be very good candidates for TDM, although prospective controlled studies are still needed. Additionally, focus is also changing from drug levels associated with efficacy to drug levels associated with toxicity. In summary, the discipline of clinical pharmacology continues to contribute valuable information to our understanding of why and how patients respond to HIV drugs.
References:
All abstracts are from 4th International Workshop on Clinical Pharmacology of HIV Therapy. March 27 - 29, 2003. Cannes, France, unless noted otherwise.
1. Penzak S, Tavel J, Acosta E, Turner M, and Masur H. Quality-control analysis of generic nevirapine formulations in the developing world: An initial report [abstract 1].
2. Maponga C, Simoyi M, Reichman R, Esch L, Slish J, and Morse G. Promoting adherence to therapy using lay community colunteers: Experience from urban Zimbabwe [abstract 2].
3. Maponga C, Simoyi M, Esch L, Catanzaro R, Hewitt R, Reichman R, and Morse G. Transitioning an established antiretroviral adherence-pharmacology unit to developing countries [abstract3].
4. Mallal S, Nolan D, Witt C, Masel G, Martin AM, Moore C, Sayer D, Castley A, Mamotte C, Maxwell D, James I, Christiansen FT. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002;359(9308):727-32.
5. Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest 1998;101(2):289-94.
6. Kim RB, Leake BF, Choo EF, Dresser GK, Kubba SV, Schwarz UI, Taylor A, Xie HG, McKinsey J, Zhou S, Lan LB, Schuetz JD, Schuetz EG, Wilkinson GR. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther 2001;70(2):189-99.
7. Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, Lamba V, Parviz F, Duncan SA, Inoue Y, Gonzalez FJ, Schuetz EG, Kim RB. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 2003;9(2):220-4.
8. Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther 2003;304(1):223-8.
9. Fellay J, Marzolini C, Meaden ER, Back DJ, Buclin T, Chave JP, Decosterd LA, Furrer H, Opravil M, Pantaleo G, Retelska D, Ruiz L, Schinkel AH, Vernazza P, Eap CB, Telenti A. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet 2002;359(9300):30-6.
10. Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A 2000;97(7):3473-8.
11. Anderson PL, Lamba J, Schuetz E, and Fletcher CV. MDR1 genotypes associated with antiviral dynamics and indinavir disposition in HIV-infected patients [abstract 4].
12. Huisman MT, Smit JW, Crommentuyn KM, Zelcer N, Wiltshire HR, Beijnen JH, Schinkel AH. Multidrug resistance protein 2 (MRP2) transports HIV protease inhibitors, and transport can be enhanced by other drugs. AIDS 2002;16(17):2295-301.
13. Kruse G, Staszewski S, Cascorbi I, Breske A, Moecklinghoff C, and Stocker H. Mutations in the drug transporter genes MDR1 and MRP2 and pharmacokinetics in patients treated with saquinavir/lopinavir [abstract 7].
14. Huisman M, Crommentuyn K, Rosing H, Beijnen J, and Schinkel A. Co-adminstration of multidrug resistance protein 2 (MRP2)-stimulators dramatically reduces saquanivir oral availability [abstract 6].
15. Camus M, Delomenie C, Faye A, Lecureur V, Gil S, and Farinotti R. Overexpression of MDR1 in placentas from HIV infected women [abstract 8].
16. Ford J, Almond L, Chandler B, Khoo S, and Back D. The effect of antiretroviral protease inhibitors on P-gp expression in CD4 and CD8 subpopulations of lymphocytes [abstract 5].
17. la Porte C, Burger D, Gyssens I, Sprenger H, and Koopmans P. Gender differences in nevirapine pharmacokinetics, fact or fiction? [abstract 10].
18. Burger D, la Porte C, van der Ende M, Miesen W, and Koopmans P.Gender-related differences in efavirenz pharmacokinetics [abstract 15].
19. Anderson PL, Kakuda TN, Kawle S, and Fletcher CV. Sex differences in zidovudine and lamivudine triphosphate concentrations in HIV-infected patients [abstract 53].
20. Poirer J, Zouai O, Meynard J, Lacombe K, Guiard-Schmid J, Girard P, and Jaillon P. Lack of effect of gender, age, weight, and body mass index on trough lopinavir plasma concentrations in HIV-experienced patients treated with Kaletra [abstract 49].
21. Chappuy H, Treluyer J, Dimet J, Rey E, Foucher M, Firtion G, Pons G, and Mandelbrot L. Maternal-fetal transfer and amniotic fluid accumulation of antiretroviral drugs in HIV-infected pregnant women [abstract 12].
22. Lorber M, Shahar E, Averbuch D, Maayan S, Burk M, Agmon-Levin N, and Schapiro J. Nelfinavir and lopinavir plasma concentrations in HIV infected patients from different origins [abstract 13].
23. Regazzi M, Villani P, Zucchi P, Cusato M, Sighinolfi L, Catania A, Guaraldi G, Calzetti C, Giacomazzi D, Stoppini L, Rossi M, Castelli P, Palvarini L, and Maserati R. Clinical Pharmacokinetics of nelfinavir and metabolite M8 in HIV/HCV co-infected patients with and without cirrhosis [abstract 14].
24. Peng J, Bertz R, Hsu A, Li J, Ashbrenner E, Grebner K, Moseley J, Reisch T, Denissen J, Cameron W, Bernstein B. Evaluation of single and multiple dose pharmacokinetics of ritonavir in subjects with mild or moderate hepatic insufficiency [abstract 16].
25. Almond L, Boffito M, Hoggard P, Bonora S, Raiteri R, Reynolds H, Khoo S, Di Perri G, and Back D. Relationship between nevirapine plasma concentration and abnormal liver function tests in a cohort of heterogeneous HIV+ patients [abstract 19].
26. Peytavin G, Meynard J, Lamotte C, Vray M, Matheron S, Morand-Joubert L, Girard P, Brun-Vezinet F, and Costagliola D. Impact of NNRTI plasma concentrations on virological response to antiretroviral therapy in HIV-1 infected NNRTI-naïve patients in ANRS 088 trial [abstract 17].
27. Garraffo R, Lavrut T, Pierre B, Durant J, and Dellamonica P. Efavirenz and nevirapine concentration-effect relationships in HIV-infected patients pharmacologically followed by routine TDM [abstract 26].
28. Peytavin G, Duval X, Lamotte C, Ecobichon J, Decamps D, Breton G, Damond F, Languet P, Leport C, and Vilde J. Indinavir hair concentrations as an indicator of indinavir long term exposure in HIV-infected patients treated with indinavir/ritonavir combination [abstract 46].
29. Duval X, Mentre F, Spire B, Chene G, Panhard X, Herson S, Massip P, Brun-Venzinet F, Raffi F, Peytavin G, and APROCO group. Early virologic response is correlated with indinavir plasma level in highly adherent HIV infected patients started on a protease inhibitor containing HAART; APROCO group [abstract 21].
30. Stevens R, Kakuda T, Bertz R, Mo H, Molla A, Rode R, and Kempf D. Inhibitory quotient of protease inhibitors using a standardized determination of IC50 [abstract 18].
31. Boffito M, Bonora S, Sinicco A, Raiteri R, Hoggard P, Khoo S, Back D, and Di Perri G. Lopinavir plasma concentrations and lipid elevation patterns in patients on lopinavir/ritonavir-containing regimens [abstract 20].
32. Valerio L, Fontas E, Garaffo R, Pradier C, Durant J, Lavrut T, and Dellamonica P. Effect of LPV trough level on the biological markers of cardiovascular risk in LPV/r treated HIV infected patients [abstract 22].
33. Raspall T, Blanco J, Lopez-Pua Y, Sarasa M, Martinez E, Biglia A, Milinkovic A, Laguno M, Leon A, Lonca M, Garcia F, Miro J, Boget T, Blanch J, Salamero M, Carne X, Gatell J, and Mallolas J. Neuropsycological disturbances and PK levels in patients receiving efavirenz: A pilot study [abstract 23].
34. Heripret L, Durant J, Clevenbergh P, Lavrut T, Dellamonica P, and Garraffo R. Absence of relationship between changes in fat distribution and antiretroviral plasma levels [abstract 24].
35. Dragovic G and Jevtovic D. Incidence of nephrolithiasis induced with indinavir plus ritonavir (boosting dose) in HIV infected patients [abstract 25].
36. Faroux S, Berhoune M, Savageon-Martre H, Timsit J, Bellenger P, Oksenhendler E, and Faure P. TDM of efavirenz in HIV-infected patients treated by Sustiva associated or not with ritonavir [abstract 37].
37. Kaul S, Damle B, Bassi K, Xie J, Gale J, Ryan K, and Hanna G. Pharmacokinetics evaluation of reduced doses of didanosine enteric coated capsules (ddI EC) in combination with tenofovir disproxil fumarate and food for a once daily antiretroviral regimen [abstract 54].
38. Lamotte C, Kirstetter M, Landman R, Lariven S, Reynes J, Katlama C, and Peytavin G. Elevated didanosine plasma concentrations in HIV-infected patients treated by a tenofovir disoproxil fumarate enteric coated ddI-containing regimen [abstract 59].
39. la Porte C, Aarnoutse R, Koopmans P, Reiss P, Lange J, Stek Jr, M, Hekster Y, and Burger D. Pharmacokinetics and food effect study of once daily indinavir/ritonavir with once daily ddI EC [abstract 11].
40. Penzak S, Shen J, Alfaro A, Remaley A, and Falloon J. Influence of low dose-ritonavir on the pharmacokinetics of the P-gp substrate digoxin [abstract 9].
41. Burger D, Schmitz K, Schneider K, Rockstroh J, and Fatkenheuer G. Pharmacokinetics of lopinavir and reduced - does indinavir as part of a salvage therapy regimen [abstract 55].
42. Tseng A, Phillips E, Antoniou T, Walker S, van Heeswijk R, and Giguere P. Steady-state pharmacokinetics and tolerability of indinavir when co-administered with lopinavir/r in antiretroviral-experienced subjects [abstract 63].
43. Taylor S, Isaac A, Rubin G, Cane P, Gibbons S, White D, Drake S, and Back D. Lopinavir/ritonavir combined with 400 mg of indinavir twice daily: pharmacokinetics in CSF, semen, and blood (The Protect Study) [abstract 68].
44. Aarnoutse R, Brinkman K, Benetucci J, Begovac J, Stek Jr, M, and Burger D. Pharmacokinetics of indinavir and low-dose ritonavir (800/100mg twice daily) combined with efavirenz in HIV-infected patients in the EASIER study [abstract 60].
45. Poirier J, Zouai O, Meynard J, Guird-Schmid J, Lacombe K, Girard P, Rozenbaum W, and Jaillon P. Lack of drug-drug interaction between lopinavir and saquinavir based on their trough plasma concentrations in highly HIV-experienced patients treated with Kaletra and Inverase [abstract 65].
46. Neff G, Tzakes A, Safdar K, and Jayaweera D. Liver transplantation in HIV, complex pharmacokinetic interactions between tacrolimus and highly active antiretroviral therapy [abstract 57].
47. Wiltshire H, McKay D, Waring S, and Aisbitt L. Predicted and observed single dose pharmacokinetics of the new broad spectrum HIV protease inhibitor, Ro334649 in healthy male volunteers [abstract 38].
48. Hoetelmans R, van der Sandt I, De Pauw M, Struble K, Peeters M, and van der Geest R. TMC114, a next generation HIV protease inhibitor: Pharmacokinetics and safety following oral administration of multiple doses with and without ritonavir in healthy volunteers [abstract 39].
49. Hoetelmans R, Baede-van Dijk P, Gruzdev B, Rakhmanova A, Doubovskaya E, Yakovlev, Peeters M, De Dier K, Struble K, and Van't Klooster G. TMC125, a next generation NNRTI: Pharmacokinetics and saaaafety following oral administration of multiple doses in antiretroviral naïve HIV-1 infected patients [abstract 40].
50. Abel S, Rosario M, van der Ryst E, Muirhead G, Edgington A, and Weissgerber G. Population pharmacokinetics of UK-427,857, a novel CCR5 antagonist, at steady-state in healthy volunteers [abstract45].
51. Raber S, Reynolds R, Hee B, Xu Y, Paxton W, Hawley P, and Amantea M. Evaluation of the pharmacokinetic drug interaction between capravirine and nelfinavir in healthy volunteers and HIV-infected patients [abstract 61].
52. Amantea M, Raber S, Pesano R, Garrett M, Ballow C, Lertratanakul A, Fredrickson J, Polan B, and Wada R. Pharmacokinetic model of capravirine, a novel NNRTI, co-administered with Kaletra in healthy and HIV-infected subjects [abstract 56].
53. Vincent I, Piketty C, Gerard L, Chazallon C, Clavel F, Calvez V, Aboulker J, Taburet A, Girard P, and the Puzzle 2 study group. Pharmacokinetic parameters of atazanavir/ritonavir when combined to tenofovir in HIV-infected patients with multiple treatment failures: a substudy of puzzle2-ANRS 107 trial [abstract 62].
54. Prelutsky D, Salvato P, and Flacon R. Pharmacokinetics of saquinavir hard gel (Inverase) when combined with atazanavir [abstract 64].
55. Charoin J, Oxley P, Gerber M, Saiedabadi N, and Kaeser B. Pharmacokinetics of Roche nelfinavir 625mg film-coated tablets and nelfinavir 250mg film-coated tablets (Viracept) are comparable [abstract 42].
56. Nieto-Cisneros L, Johnson M, Horban A, Arasteh K, Gonzales-Garcia J, Proll S, Foreman R, and Mueller T. Investigation of the gastrointestinal-tolerability and pharmacokinetics of the Roche nelfinavir 625mg film-coated tablets in comparison with nelfinavir 250mg film-coated tablets (Viracept) in HIV-infected patients [abstract 43].
57. Boffito M, Hoggard P, Lindup E, Tija J, Bonora S, Sinicco A, Khoo S, Di Perri G, and Back D. Lopinavir protein binding in vivo through the 12 hour dosing interval: ultrafiltration versus equilibrium dialysis [abstract 66].
58. Au S, Gates A, Blaschke T, Robbins R, and Aweeka F. Variability on alpha-1 acid glycoprotein and its impact on nelfinavir free fraction in HIV-infected subjects [abstract 67].
59. Grassi J, Becher F, Pruvost A, Sclemmer D, Creminon C, Benech H, Boutet V, and Goujard C. New lights on the intracellular pharmacology of NRTIs in HIV-infected patients [abstract 52].
|
|
| |
|
|
|
|
|