| |
Future Therapy of Hepatitis C
|
| |
| |
John G. McHutchison, MD Duke Clinical Research Institute, Durham, NC Hepatology, November 2002
Editorial note from Jules Levin: Although this article was written last year, it is a good review of potential new therapies for HCV, there may be updates for research discussed in the article. In particular, at the November 2003 AASLD liver meeting advances were announced for several HCV protease inhibitors: VX-450 and Schering 7 are advancing into the next phase of clinical study. Future development of BILN 2061 remains uncertain, but it appears that backup compounds are behind it. You can read reports from the AASLD meeting at: http://www.natap.org/2003/AASLD/ndxAASLD.htm
ABSTRACT. Currently available therapies for the treatment of chronic hepatitis C are effective in half of patients, but are expensive, often poorly tolerated, and unsuitable for certain patient populations. The ideal therapy would be highly effective, orally bioavailable, have minimal side effects, be cost effective, and suitable for the majority of patients with hepatitis C.
Recent advances in understanding the replication cycle of hepatitis C virus (HCV) and structural, crystallographic definitions of components of the viral polyprotein have improved the prospects for development of novel therapies. The lack of a small animal model of HCV infection continues to hamper progress in the preclinical evaluation of new antivirals and vaccines. Strategies to enhance response to current therapies include the development of novel interferons and delivery systems, nucleoside analogues that have reduced hemolysis compared with ribavirin, inosine 5' monophosphate dehydrogenase inhibitors, and other immunomodulators that are being evaluated as adjunctive therapy to interferon-based regimens.
Compounds in preclinical or early phase human trials include small molecules that inhibit virus specific enzymes (such as the serine proteases, RNA polymerase and helicase), or those that prevent translation initiation (such as antisense molecules and ribozymes).
Antifibrotic agents are also being developed in an attempt to prevent disease progression in patients in whom HCV RNA cannot be eradicated. While the advent of these newer compounds represent an exciting phase in the treatment of HCV, their safety and efficacy need to be established. Most of these newer therapies are unlikely to be available for routine clinical use in the next 3 to 5 years.
BACKGROUND
In recent years, there have been significant advances in our understanding of the molecular virology of the hepatitis C virus (HCV) as well as in the means of treatment of chronic hepatitis C. Current treatment options now provide effective therapy in over one half of patients. However, treatment is costly, prolonged (6 or 12 months), not suitable for all patients, associated with significant morbidity, and requires a substantial commitment from patients and health care staff alike. Furthermore, treatment options are limited for patients with advanced liver disease or those with contraindications to current therapy. As an increasing number of chronic hepatitis C patients are treated, the number of nonresponders to currently available therapies will also rise, resulting in a larger group of patients with limited alternative therapeutic options.
The lack of an effective cell culture system and small animal model for HCV has slowed drug and vaccine development. Improvements in our knowledge of the function of the viral genome, replication intermediates, encoded proteins, and their interaction with the host immune response may help identify targets for future therapy. The ideal therapy for patients with chronic hepatitis C would be highly effective, orally bioavailable, suitable for the majority of patients, without major side effects, and cost effective. An ideal therapy is unlikely to be available in the near future, but a number of novel and potentially useful compounds are currently under development that may significantly improve current options for therapy.
An exhaustive review of all potential future strategies is beyond the scope of this article. Newer therapies that are currently or soon to be in clinical development in chronic hepatitis C patients, and as such, may be available in the next 3 to 5 years, will be discussed.
LIMITATIONSTO DRUG DEVELOPMENT
Despite the development of numerous in vitro diagnostic enzyme assays based on HCV genome targets, the absence of a stable cell-based system supporting HCV replication has hampered drug development. In the interim, the use of surrogate virus models closely related to HCV, such as the bovine virus diarrheal virus and the tamarin GB virus-B, provides alternate approaches.
However, biological activity against these related viruses may not translate into antiviral activity in human HCV infection. At this stage the only well-established animal model supporting HCV replication is the chimpanzee, and work with this endangered species is expensive and restricted to only a few centers. The recent description of a complex transgenic mouse model that allows HCV replication may provide a suitable alternative small animal model in the future. The development of subgenomic replicons containing HCV nonstructural proteins that replicate in human hepatoma cells now provides a system to test or screen candidate drugs that target the nonstructural gene products. However, these subgenomic replicons do not produce infectious virions, limiting the ability to study cell-to-cell viral transmission.
The genetic heterogeneity of HCV suggests that future compounds should display activity against a broad range of HCV genotypes, subtypes, and quasispecies. Furthermore, experience with antiviral therapy for human immunodeficiency virus 1 and hepatitis B infected patients indicates that the emergence of resistance will be an important issue. This suggests that combination therapy aimed at several viral and/or host immune targets will probably become the future approach to treatment to reduce or eliminate drug resistance.
STRATEGIES TO ENHANCE RESPONSE TO CURRENT THERAPIES
Alternative interferons or methods of delivery
Novel delivery systems under development to improve the pharmacokinetic profile of interferons include disposable infusion pumps, conjugation with albumin, controlled release formulations for parenteral use (including implantable sustained release devices), liposome encapsulation, and polyaminoacid-based oral delivery systems. Alternative parenteral type-I interferons are also currently being evaluated, as are potential oral interferon-inducing compounds. While some of these alternate interferons and delivery systems are in clinical development, their clinical use, indications, efficacy, and side-effect profile compared with current type-1 interferons must be established further.
Ribavirin-like drugs
The precise mechanisms of action of ribavirin against HCV are unknown, but are likely pleiotropic and may involve a combination of direct inhibition of the HCV NS5B polymerase, immunomodulation through inosine 5'-monophosphate dehydrogenase (IMPDH) inhibition and other pathways, direct RNA mutagenic activity, and promotion of the host immune response towards a Th-1 profile. Levovirin, a second generation L-isomer of ribavirin, retains the immunomodulatory activity of ribavirin by enhancing the Th-1 cytokine response in vitro. The drug is also associated with lesser degrees of hemolytic anemia compared with ribavirin, presumably because it is not converted in erythrocytes to mono-, di- and tri-phosphate intermediates. Phase I dose-ranging studies in healthy volunteers indicate the drug is well tolerated. Further clinical trials in combination with peginterferons are warranted.
Viramidine is a ribavirin “pro-drug” that is rapidly converted into ribavirin in vivo. The compound has at least a 3-fold longer residence time in the liver, and produces less hemolysis in animals because of its reduced relative uptake into erythrocytes compared with ribavirin.7,8 An initial phase I study indicated an acceptable safety profile and further phase II development of viramidine in combination with interferon is under way.
IMPDH inhibitors
One of the antiviral mechanisms of action of ribavirin is inhibition of IMPDH, a rate-limiting enzyme involved in de novo guanine nucleotide synthesis. Depletion of intracellular reservoirs of guanine nucleotides may inhibit viral replication.
VX-497 is an orally bioavailable small molecule that is a specific and potent inhibitor of IMPDH. The drug has a broad antiviral spectrum in vitro, including activity against bovine virus diarrheal virus. In an HCV replicon system VX-497 is inhibitory and has additive inhibitory effects on viral replication when combined with interferon and ribavirin. In a small safety and pharmacokinetic study, VX-497 monotherapy was well tolerated, a reduction in alanine aminotransferase values was observed, but there was no effect on HCV RNA concentrations (similar to ribavirin monotherapy). A further 4-week placebo-controlled study in 53 treatment naive patients evaluating VX-497 in combination with interferon-alfa indicated the drug was well tolerated, with no significant changes in hemoglobin levels compared with the placebo group. At 4 weeks the antiviral effects of the VX-497 combination therapy were similar to those observed with interferon monotherapy. Treatment studies of longer duration are required to better define the antiviral activity and sustained response rates of this drug in combination with interferon alfa. Further development of more potent IMPDH inhibitors is in progress. Mycophenylate mofetil, which also inhibits IMPDH, is also currently being evaluated in a large clinical study in combination with peginterferon in patients that are nonresponders to prior interferon alfa and ribavirin therapy.
Histamine
Histamine dihydrochloride binds histamine type 2 receptors on intrahepatic phagocytic cells and reduces generation of free radicals through inhibition of the NADPH-oxidase pathway. Histamine dihydrochloride also exhibits a variety of immunomodulatory activities via natural killer cells and T lymphocytes. Phase II studies have evaluated a range of doses of histamine dihydrochloride in combination with interferon alone, or interferon and ribavirin, and suggest some benefit in terms of virological response. The lack of a control group makes interpretation of these preliminary results difficult. An ongoing randomized multicenter open-label European study evaluating the efficacy of histamine in combination with peginterferon and ribavirin is in progress.
Thymosin alpha 1
Thymosin alpha 1 is a synthetic 28 amino acid nonglycosylated peptide derivative of a purified thymosin fraction of thymus gland extracts. It has been shown to promote T-cell maturation and natural killer cell activity, and stimulate production of interferon gamma, interleukin (IL)-2, and IL-3, in addition to other immunomodulatory actions. Three pilot studies have suggested thymosin alpha 1 is safe when used in combination with interferon alfa. Unfortunately, the sustained response rate was not available in 1 of these studies, and the study designs precluded interpretation of the benefits of the combination. At present, 2 double-blind, multicenter trials, enrolling approximately 1,000 patients, are evaluating the efficacy of thymosin alpha 1 and peginterferon in cirrhotic and noncirrhotic patients that have not responded to prior therapy with interferon alfa, alone or in combination with ribavirin.
Amantadine
Amantadine, a tricyclic amine, has antiviral activity against a broad range of viruses, and like its related compound rimantadine, has been used to prevent and limit influenza A infection. Furthermore, amantadine is safe, well tolerated, and relatively inexpensive. After an initial positive report of the efficacy of amantadine in patients with chronic hepatitis C, a series of clinical trials have evaluated amantadine alone or in combination with interferon alfa with or without ribavirin. In vitro studies to date have indicated that amantadine has little or no direct inhibitory effects on HCV replication. Four of 5 randomized controlled trials in previously untreated patients with chronic hepatitis C have reported a higher sustained virological response rate when interferon alfa is combined with amantadine compared with interferon monotherapy. A meta-analysis of 5 studies including 924 patients indicated a higher virological response rate for patients receiving both drugs compared with interferon alfa alone (22.4% vs. 16.6%; P < .03; S. Zeuzem, personal communication, June, 2002). Further randomized studies evaluating “triple therapy” in treatment-naive patients using peginterferon, ribavirin, and amantadine are now in progress. Whether amantadine in combination with interferon alfa and ribavirin enhances sustained virological response rates in prior interferon alfa monotherapy nonresponders has been evaluated in 2 studies with conflicting results. Likewise, the role of amantadine in treating interferon alfa and ribavirin nonresponders is unknown and will require further prospective evaluation.
NEW STRATEGIES: MOLECULAR-BASED THERAPY
The HCV genome
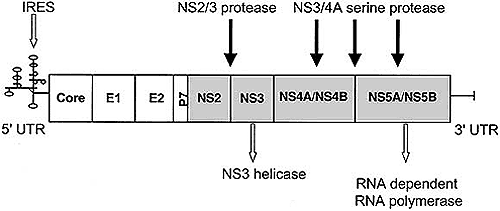
Hepatitis C virus is an enveloped positive-sense RNA virus, the genome consisting of a single open reading frame of approximately 9.6 kilobases, flanked by 5' and 3' untranslated regions that are necessary for translation and replication of the viral RNA. The 5' untranslated region contains an internal ribozyme entry site (IRES) that initiates translation of a 3,000 amino acid polyprotein, which is subsequently processed by viral and host proteinases into 10 mature structural (core, E1, E2, and p7) and nonstructural HCV proteins (NS2, NS3, NS4A, NS5A, and NS5B) (Fig. 1).
Fig. 1. Schematic representation of the HCV genome and sites for current drug development. Unshaded regions indicate the major structural proteins: core (c), envelope (E1 and E2), and P7. The shaded region indicates the nonstructural proteins NS2, NS3, NS4A/NS4B, and NS5A/NS5B. At the 5' untranslated region resides the internal ribosomal entry site (IRES), which is highly conserved and represents a site for development of translation inhibitors such as antisense oligonucleotides and ribozymes. NS3 encodes for a specific helicase and the NS5A region encodes for an RNA-dependent RNA polymerase enzyme, both important in viral replication (open arrows). These also represent sites for the development of specific viral enzyme inhibitors. Other potential enzyme targets include the HCV specific proteases (NS2/3 and NS3/4). These enzymes are involved in processing the viral polyprotein at specific sites as indicated (closed arrows).
|
|
| |
| |
 |
|
| |
| |
The structural proteins are cleaved by host-signal peptidases, and the nonstructural proteins are generated largely by HCV-encoded proteases. These nonstructural proteins encode several key enzymes involved in viral replication. Because of their specificity for HCV, many of these enzymes may be ideal drug targets.
NS2/3 protease
The NS2/3 protease is a zinc-dependent, metalloprotease that cleaves the nonstructural proteins between the NS2 and NS3 polypeptides. This region overlaps with a serine protease at the N-terminal end of NS3, which is dependent on His952 and Cys993 residues for proteolytic activity. Further characterization of the structure and function of this protease may lead to the development of specific NS2/3 protease inhibitors.2
NS3 protease
NS3 encodes a multifunctional protein that contains a serine protease (NS3 protease) in the N-terminus. The NS3 protease requires the relatively small NS4A sequence as a co-factor for proteolytic activity and mediates essential polyprotein processing through cleavage of junctions of NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B. The crystalline structure of the NS3 protease has been defined and studied extensively as a potential target for antiviral activity. The substrate-binding site, the zinc co-factor that provides structural support to the peptide, and the interaction between NS3 and NS4A are 3 possible targets for drug development. However, the NS3 protease binding site has a poorly structured and shallow substrate-binding cleft, which has been challenging to the development of NS3-specific HCV protease inhibitors. Furthermore, compounds that target the zinc co-factor are likely to be nonspecific. Finally, the strong bonds between NS3 and NS4A also pose difficulty in developing inhibitors to counteract their association. Despite these barriers, a number of substrate-based inhibitors with reactive moieties have been developed including boronic acids, -ketoamides, and hydrazinoureas. In addition, small non-peptidic molecules that inhibit NS3 protease in vitro have been described. The first early phase clinical trials with specific NS3 inhibitors are now being planned or are currently ongoing (Table 1).
NS3 helicase
The NS3 helicase mediates separation of duplex nucleotides through an unwinding mechanism important in viral replication. Energy for this process is derived from the hydrolysis of nucleoside triphosphates, suggesting this enzyme has nucleoside triphosphate activity. The crystalline structure of this helicase has also been described as a unique Y-shaped protein consisting of 3 linked domains separated by distinctive clefts. These features provide potential targets for developing compounds that inhibit either or both the RNA and nucleoside triphosphate binding sites. Newer HCV NS3 helicase inhibitors being developed include thiodiazonium compounds that are active against nucleoside triphosphate binding sites and substituted pyrimidines that affect helicase mediated unwinding. At this stage, only 1 compound (VP-50406; Viropharma Inc, Wyeth-Ayerst Laboratories, Collegeville, PA) has been in human clinical trials. However, preliminary results suggested that sufficient plasma drug concentrations were not achieved to obtain adequate antiviral activity. Further investigation of this compound has been halted in favor of the development of newer classes of helicase inhibitors.
NS5B RNA-dependent RNA polymerase
The NS5B region of the HCV genome encodes an RNA-dependent RNA polymerase enzyme. Crystallography studies of NS5B have shown a globular structure that resembles a donut-like flat sphere and consists of 3 closely interacting subdomains, termed fingers, palm, and thumbs. These subdomains completely encircle the active site, forming a cavity that is preserved during polymerization. This large binding cleft provides an attractive target for drug development, and further co-crystallization studies may lead to the discovery of a new class of inhibitors. NS5B is highly conserved across HCV genotypes and is virus specific. These features are predictive that drugs with broad antiviral activity and minimal side effects might be developed. JTK-003 (Akros Pharma Inc., Princeton, NJ) is an RNA-dependent RNA polymerase inhibitor that has been found to be well tolerated in phase I dose-ranging studies. Recently, a phase Ib multicenter, placebo-controlled, ascending-dose clinical study of JTK-003 has been initiated in HCV genotype 1-infected patients who were refractory to interferon-based therapy (Table 1). Other compounds, such as the rhodanine, diketobutanoic acid, and barbituric acid derivatives are also in preclinical evaluation.
Cell entry
The mechanism of how HCV enters cells remains unknown. One possibility is endocytosis of HCV mediated through the low-density lipoprotein receptor. Alternatively, interaction of the HCV envelope proteins with CD81 (a tetraspanin that is expressed on the surface of hepatocytes) may be involved. As such, both these events may represent potential host targets for drug development. Further characterization of the interaction between the HCV envelope proteins E2 and the conformational binding characteristics of the CD81 receptor may provide a novel approach to preventing HCV entry into hepatocytes. Receptors involved in cell tropism for other extrahepatic HCV reservoirs have yet to be characterized.
Internal ribosome entry site
The HCV IRES is a highly conserved area located within the 5' untranslated region of the genome. The IRES is composed of 4 domains and forms a binary complex with the 40S ribosome subunit, initiating translation of the viral genome.13 Domain IV consists of a small stem-loop containing the polyprotein start codon. Sequences immediately downstream of this AUG initiator are important in regulating RNA translation and serve as potential targets for small molecule inhibitors. However, 40S ribosomes also have other contact sites on domains II and III, and thus compounds with high affinity and multiple sites of action on the IRES may be required. Inhibitors of HCV translation must also bind the HCV IRES with high specificity to avoid interaction with cellular RNA.
Antisense oligonucleotides
Antisense oligodeoxynucleotides are short DNA or RNA sequences specifically designed to bind to a target mRNA. The resulting hybrid is subsequently degraded by the cellular enzyme Rnase H, thus preventing translation of disease-causing proteins. Phosphorothioate modifications also enable these compounds to resist cellular endonucleases. Several oligonucleotides have been designed to bind the HCV IRES, and are effective in reducing RNA translation in in vitro gene expression model systems. ISIS-14803 (ISIS Pharmaceuticals Inc, Carlsbad, CA) is a 20-base phosphorothioate oligonucleotide that is complementary to the initiation codon for polyprotein translation in the HCV IRES, and is under evaluation in phase I/II trials. A small initial 4-week dose-escalation study of predominantly genotype 1-infected patients that were nonresponders to interferon-based regimens, indicated that ISIS-14803 was well tolerated and resulted in significant reductions of greater than 1.0 log10 units in HCV RNA levels in some patients. Some of these responses were associated with transient asymptomatic elevations in alanine aminotransferase levels (approximately 10-fold). The safety and efficacy of this compound are presently being evaluated in phase II 12-week treatment trials. Potential limitations of this technology include the possible development of resistance, poor binding affinity of the oligonucleotide to viral RNA, and the dependence on adequate cellular RNAse H to disrupt translation.
Ribozymes
Ribozymes are catalytic RNA molecules that cleave specific RNA sequences. Variable flanking sequences complementary to the target RNA determine their specificity. Modified nucleotides and phosphorothioate linkages enable the ribozyme molecule to resist host nuclease degradation. An HCV specific ribozyme designed to cleave the HCV IRES has been developed (Heptazyme [RPI.13919]; RPI, Boulder, CO) Small animal studies have indicated distribution to the liver following parenteral administration. Chimeric cell-culture studies have also shown that this ribozyme inhibits viral replication in a dose-dependent fashion, and this effect is potentiated by interferon. Phase I trials of RPI.13919 indicated the drug was safe. A subsequent phase II dose-escalation trial assessing the safety and efficacy of this ribozyme in combination with interferon alfa has been initiated. While a reduction in serum HCV RNA was observed in some patients, dosing has recently been discontinued because of primate toxicology findings. Recruitment into the trial has been deferred until resolution of this issue.
While these newer small-molecule approaches mentioned here provide hope and excitement for the treatment of HCV-infected patients, further studies of these compounds will be necessary to determine safety and efficacy, clinical end-points, effect on liver histology, mechanisms of antiviral action, and to evaluate whether these agents will need to be administered in combination with available interferon-based therapies.
THERAPEUTIC STRATEGIES TO REDUCE HEPATIC INJURY
Although the primary aim of therapy in chronic hepatitis C is long-lasting viral eradication, secondary end-points of reducing liver inflammation and preventing fibrosis progression are also important, particularly for patients who have contraindications to current therapies or who fail to respond. The activation of hepatic stellate cells in response to injury is regulated by several soluble paracrine factors such as growth factors, cytokines, and chemokines. These transform the quiescent hepatic stellate cells into a myofibroblastic phenotype, leading to deposition of a fibrotic matrix in the hepatic sinusoids.20 Therapeutic strategies to limit fibrosis progression include removing the source of injury (achieved through eradication of HCV), blocking hepatic stellate cell activation and collagen deposition, and increasing matrix degradation. Although several antifibrotic compounds have been developed, few have been evaluated in clinical trials in patients with chronic hepatitis C.
Interferon gamma
Interferon gamma is produced by activated T lymphocytes and natural killer cells, and has many reported antimicrobial, antiproliferative, and antifibrotic activities. Interferon gamma downregulates transforming growth factor and decreases hepatic stellate cell activation and proliferation. Additionally, interferon gamma has recently been shown to inhibit protein synthesis and RNA replication in subgenomic HCV replicon constructs. Based on these observations, it seems logical that interferon gamma might both decrease HCV RNA levels and prevent fibrosis in chronic hepatitis C. A preliminary study of gamma interferon (combined with corticosteroids) in patients with idiopathic pulmonary fibrosis suggested improvement in markers of pulmonary function. A phase II, randomized, double-blind, multicenter trial of 450 patients is in progress to test the hypothesis that recombinant interferon gamma-1b (Intermune Pharmaceuticals Inc., Brisbane, CA) improves histological fibrosis in chronic hepatitis C patients with advanced fibrosis.
Other strategies to modulate host immune responses
Augmenting the host cellular immune response to HCV infection may provide several other therapeutic options for therapy. The CD4+ Th-1 response is an integral part of cell-mediated immunity, promoting secretion of proinflammatory cytokines such as IL-2, interferon gamma, and tumor necrosis factor-. The Th-1 response is counterbalanced by the Th-2 response, which is active in both cellular and humoral immunity, and is mediated through secretion of IL-4, IL-5, and IL-10. Attempts to redirect the host immune response towards a Th-1 profile have met with limited success to date.
Interleukin-12
Immunomodulatory cytokines such as recombinant human interleukin-12 (IL-12) promote cell-mediated immunity by stimulating Th-1 responses and secretion of interferon gamma from cytotoxic T lymphocytes and natural killer cells. Results from a phase II randomized, controlled, multicenter trial of IL-12 in 225 interferon or interferon and ribavirin nonresponder patients indicated minimal efficacy in terms of virological response or histological activity, but substantial toxicity. Thus, further development of IL-12 for chronic hepatitis C seems unlikely.
Interleukin-10
In patients with chronic hepatitis C, IL-10 is produced by CD4+ Th-2 cells and acts to down-regulate the inflammatory response. Although expression of IL-10 increases during hepatic stellate cell activation, progressive liver injury in hepatitis C leads to lower levels of this cytokine, indicating an antifibrogenic role. IL-10 was reported to reduce hepatic fibrosis and serum aminotransferase levels in small pilot trials of patients with hepatitis C who had not responded to combination therapy. However, a follow-up study of patients with advanced fibrosis treated with IL-10 for 12 months failed to show a significant benefit for this cytokine in terms of fibrosis as a histological end-point.23 Furthermore, HCV RNA levels increased by as much as 3-fold during the 1-year of therapy. It seems unlikely that this compound will undergo further clinical evaluation in chronic hepatitis C patients.
Therapeutic vaccines
The lack of a vigorous and multispecific cytotoxic T-lymphocyte response in the early phase of acute HCV infection is believed to be important in allowing viral persistence and development of chronic infection. In animal models, vaccination with envelope glycoprotein subunits gpE1 and gpE2 may prevent chronic infection following challenge with homologous strains of HCV. Preliminary results of a phase II placebo-controlled human study of an E1 therapeutic vaccine (Innogenetics, Gent, Belgium) administered to patients with hepatitis C suggest that E1 antibody levels and specific T-cell responses can be readily induced. The effects of E1 vaccination on HCV RNA concentrations, serum aminotransferase levels, and liver disease severity are unknown.
The passive transfer of anti-HCV immunoglobulin decreases HCV RNA levels in primates with acute and chronic HCV infection. Dose-ranging phase I/II studies are planned using hepatitis C immune globulin (Civacir; NABI Inc., Rockville, MD) in a randomized, controlled trial to evaluate whether this therapy prevents graft reinfection or injury following orthotopic liver transplantation in HCV-infected patients.24 Safety, antibody levels, and serum and liver HCV RNA concentrations will be evaluated as part of this study.
Conclusions TOP
In the immediate future, therapies for patients with hepatitis C will rely on interferon-based regimens. The utility of alternative interferon preparations and delivery methods, and other ribavirin-like drugs, needs to be established, but they are likely only to supplement current therapies. These newer antiviral agents will need to show synergistic effects with current therapy and to be safe and enhance sustained response rates.
While many other exciting future strategies are presently in early developmental stages, it will take several years to determine their safety, short- and long-term efficacy, and appropriate clinical settings for use (Table 1). As in the human immunodeficiency virus 1 treatment setting, multiple drug regimens attacking several targets will almost certainly be required to prevent or diminish viral resistance. For all these reasons, these newer anti-viral and molecular-based approaches (if effective) will not be available for routine clinical use in the next 3 to 5 years.
|
|
| |
| |
|
|
|