 |
|
|
| |
Tenofovir vs d4T + EFV/3TC, study 903: 144 weeks in men & women
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
"Efficacy and Safety of Tenofovir DF vs Stavudine in Combination Therapy in Antiretroviral-Naive Patients": women's substudy
A 3-Year Randomized Trial
JAMA -- Jnl of the American Medical Assn
Vol. 292 No. 2, July 14, 2004
Joel E. Gallant, MD, MPH; Schlomo Staszewski, MD; Anton L. Pozniak, MD; Edwin DeJesus, MD; Jamal M. A. H. Suleiman, MD; Michael D. Miller, PhD; Dion F. Coakley, PharmD; Biao Lu, PhD; John J. Toole, MD, PhD; Andrew K. Cheng, MD, PhD; for the 903 Study Group
ABSTRACT
Context Tenofovir disoproxil fumarate (DF) is a once-daily nucleotide analogue reverse transcriptase inhibitor.
Objective To evaluate the efficacy and safety of tenofovir DF compared with stavudine in antiretroviral-naive patients.
Design, Setting, and Participants A prospective, randomized, double-blind study conducted at 81 centers in the United States, South America, and Europe from June 9, 2000, to January 30, 2004. A total of 753 patients infected with HIV who were antiretroviral naive were screened and 602 patients entered the study.
Baseline demographic and laboratory characteristics were comparable between the 2 groups: 75% men; 26% women; 64% white; 20% Black; 7% Hispanic. HIV RNA 4.91 log. Mean HIV RNA 81,300 copies/ml. 45% >100,000 c/ml. CD 280. CD4s <200: 39%; CD4 <50 15%.
Intervention Patients were randomized to receive either tenofovir DF (n = 299) or stavudine (n = 303), with placebo, in combination with lamivudine and efavirenz.
Main Outcome Measure Proportion of patients with HIV RNA levels of less than 400 copies/mL at week 48.
Results In the primary intent-to-treat analysis in which patients with missing data or who added or switched antiretroviral medications before week 48 were considered as failures, the proportion of patients with HIV RNA of less than 400 copies/mL at week 48 was 239 (80%) of 299 in patients receiving tenofovir DF and 253 (84%) of 301 in patients receiving stavudine (95% confidence interval, --10.4% to 1.5%), exceeding the predefined --10% limit for equivalence.
However, equivalence was demonstrated in the secondary analyses (HIV RNA <50 copies/mL) at week 48 and through 144 weeks.
Virologic failure was associated most frequently with efavirenz and lamivudine resistance.
Through 144 weeks, the study regimen permanent discontinuation rate was 82 (27%) of 299 in the tenofovir DF group and 100 (33%) of 301 in the stavudine group.
Through 144 weeks, the K65R mutation emerged in 8 and 2 patients in the tenofovir DF and stavudine groups, respectively (P = .06).
A more favorable mean change from baseline in fasting lipid profile was noted in the tenofovir DF group at week 144: for triglyceride levels (+1 mg/dL for tenofovir DF [n = 170] vs +134 mg/dL for stavudine [n = 162], P<.001), total cholesterol (+30 mg/dL [n = 170] vs +58 mg/dL [n = 162], P<.001), direct low-density lipoprotein cholesterol (+14 mg/dL [n = 169] vs +26 mg/dL [n = 161], P<.001), and high-density lipoprotein cholesterol (+9 mg/dL [n = 168] vs +6 mg/dL [n = 154], P = .003).
There were no significant differences between groups in fasting lipid profile at baseline. Investigators were allowed to prescribe lipid-lowering agents (consisting of a statin and/or a fibrate derivative) based on clinical judgment during the course of the study. A Kaplan-Meier analysis of time to use of first lipid-lowering agent (patients on lipid-lowering agents at baseline were excluded) showed that through 144 weeks, 38 of 301 patients (16%; 95% CI, 11.3%-20.7%) receiving stavudine initiated a lipid-lowering agent compared with 11 of 294 patients (5%; 95% CI, 2.0%-7.4%) receiving tenofovir DF (P<.001).
Investigator-reported lipodystrophy was less common in the tenofovir DF group compared with the stavudine group (9 [3%] of 299 vs 58 [19%] of 301, P<.001).
Toxicities that have been attributed to mitochondrial toxicity (peripheral neuropathy, lipodystrophy, and lactic acidosis) were reported in 100 patients, 83 (28%) of 301 in the stavudine group and 17 (6%) of 299 in the tenofovir DF group (P<.001). Neuropathy was observed in 31 (10%) of 301 and 9 (3%) of 299 patients in the stavudine and tenofovir DF groups, respectively (P<.001).
Using whole body DXA, significantly less total limb fat was observed in the stavudine group at week 96 (7.9 kg tenofovir DF [n = 128] vs 5.0 kg stavudine [n = 134], P<.001) and week 144 (8.6 kg tenofovir DF [n = 115] vs 4.5 kg stavudine [n = 117], P<.001). Investigator-defined lactic acidosis occurred in 3 patients, all of whom were in the stavudine group.
The number of bone fractures and the renal safety profile were similar between the 2 groups.
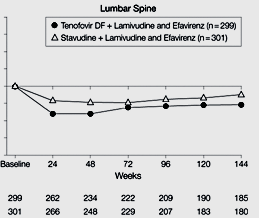
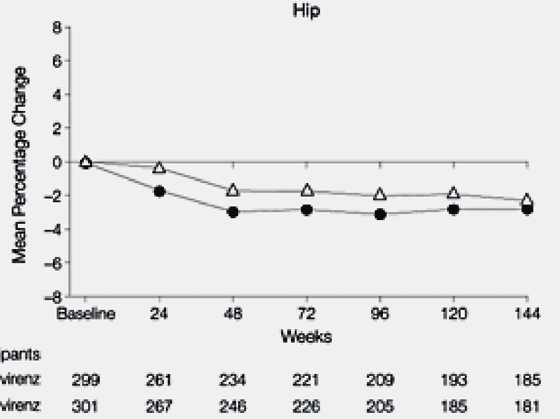
At week 144, a greater mean percentage decrease from baseline in bone mineral density was observed at the lumbar spine in the tenofovir DF group (--2.2% tenofovir DF vs --1.0% stavudine, P = .001) but similar changes were observed at the hip (--2.8% tenofovir DF vs --2.4% stavudine, P = .06). Notably, these decreases occurred through weeks 24 to 48 and stabilized through week 144 (Figure 3). Sixteen patients (11 in the stavudine group and 5 in the tenofovir DF group) developed bone fractures through 144 weeks. Nearly all fractures were related to trauma, except for a vertebral compression fracture for 1 patient in the stavudine group.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
Through 144 weeks, the renal safety profile was similar between the 2 groups (Table 5). Two patients in each group developed a creatinine level of more than 2.0 mg/dL (>176.8 µmol/L), while hypophosphatemia (<2.0 mg/dL [<176.8 µmol/L]) was observed in 10 patients receiving tenofovir DF and 8 patients receiving stavudine. The incidence of proteinuria and/or glycosuria was similar between the two groups. No patient developed Fanconi syndrome or discontinued from study due to tenofovir DF--related renal abnormalities. The renal safety profile associated with these drugs is addressed in more detail in other analyses.
A total of 20 patients (11 of 299 in the tenofovir DF group and 9 of 301 in the stavudine group, P = .40) reported 21 category C AIDS-defining conditions (based on the Centers for Disease Control and Prevention 1993 revised guidelines37) at least 30 days after the first dose of study drugs. These category C conditions were disseminated Mycobacterium avium complex, tuberculosis, cytomegalovirus retinitis, Pneumocystis jiroveci pneumonia, progressive multifocal leukoencephalopathy, cryptococcosis, cryptosporidiosis, toxoplasmosis, Kaposi sarcoma, esophageal candidiasis, chronic herpes simplex, and recurrent pneumonia.
Conclusions Through 144 weeks, the combination of tenofovir DF, lamivudine, and efavirenz was highly effective and comparable with stavudine, lamivudine, and efavirenz in antiretroviral-naive patients. However, tenofovir DF appeared to be associated with better lipid profiles and less lipodystrophy.
WOMEN'S SUBSTUDY: 144 weeks results
Presented at Bangkok IAC
N=155 women (26% of study population
Mean age: 35
Weight: 65 kg
51-59% white
28-34% black
3% Hispanic
10-13% other
mean HIV RNA 4.7 log
mean CD4 288
DISPOSITION
Permenant withdrawal prior to wk 144: 26-27%
Reasons: 13% noncompliance in TDF arm vs 0% in d4T arm
Withdrew consent: 3% TDF vs 12% d4T. Otherwise no differences between arms.
PROPORTION WITH HIV RNA <50 c/ml:
67% TDF, 64% d4T. For men: 75% TDF, 71% d4T.
CD4 INCREASE: 314 TDF, 320 d4T. For men: 344 TDF, 272 d4T.
GRADE 3/4 ADVERSE EVENTS THROUGH WK 144:
Any grade 3/4 event: 26% TDF, 17% d4T. For men: 27% TDF, 28% d4T.
GRADE 3/4 LAB ABNORMALITIES THROUGH WK 144:
Ant grade 3/4 event: 29% TDF, 41% d4T. For men: 39% TDF, 43% d4T
TG: 0% TDF, 3% d4T. For men: 4% TDF, 17% d4T
Amylase: 5% TDF, 12% d4T. For men: 11% TDF, 6% d4T
Creatinine kinase: 6% TDF, 3% d4T. For men: 14% TDF, 15% d4T
AST: 3% TDF, 5% d4T. For men: 6% TDF, 7% d4T
ALT: 3% TDF, 4% d4T. For men: 5% TDF, 4% d4T
Neutropenia: 5% TDF, 1% d4T. For men: 3% TDF, <1% d4T
MEAN CHANGE FROM BASELINE IN FASTING LIPIDS THROUGH WK 144
Total chol: + 23 TDF, + 58 d4T. For men: +33 TDF, +58 d4T
TG: -4 TDF, +71 d4T. For men: +2 TDF, +151 d4T
LDL-C: +4 TDF, +34 d4T. For men: +18 TDF, +24 d4T
HDL-C: +11 TDF, +8 d4T. For men: +9 TDF, +8 d4T
LIMB FAT
TDF-Week 96: 10.9 vs Week 144: 11.1. For men: looks like 6.8 wk 96 vs 7.6 wk 144
D4T- week 96: 7.8 vs week 144 7.1. For men: d4T- 4.0 wk 96 vs 3.5 wk 144
(p<0.001)
INCIDENCE OF GRADED SERUM CREATININE AND PHOSPHOROUS THROUGH WEEK 144
GRADED SERUM CREATININE (mg/dL) (mmol/L)
1 (>=0.5 increase from baseline) (>44 increase from baseline
3% TDF, 4% d4T
2 (3.1-3.0) (188-265): 0% TDF, 0% d4T
3(3.1-6.0) (265-530): 0% TDF, 0% d4T
4 (>6.0) (>530): 0% TDF, 0% d4T
SAME FOR MEN IN ALL OF ABOVE
GRADED SERUM PHOSPHOROUS (mg/dL) (mmol/L)
1 (2.0-<2.2) (0.65-<0.71): 3% TDF, 1% d4T. For men: 5% TDF, 4% d4T
2 (1.5-1.9) (0.48-0.64): 1% TDF, 0% d4T. For men: 4% TDF, 3% d4T
3 (1.0-1.4 (0.32-0.47): 1% TDF, 0% d4T. same for men
4 (<1.0) (<0.32): 0% TDF, 0% d4T. same for men
MEAN % CHANGE IN HIP & SPINE BMD
Mean decreases from baseline in bone mineral density were higher in women on the TDF arm: no bone fractures were reported in women. By visual judging of graph it appeared to be decrease of --2% for women in TDF arm vs 0% in d4T arm at week 144. the difference between the arms started to appear at wk 24 (p=0.029 & 0.032, respectively, Spine & HIP BMD). For men there was a difference reported in Spine BMD with TDF performing better than d4T (p=0.008), but no difference for men between TDF & d4T for hip BMD (p=0.343).
Overall in the study, there were 16 fractures reported through week 144: 11 were on d4T vs 6 on TDF; 15 were trauma related while one (d4T) was reported as vertebral compression fracture.
AUTHOR CONCLUSIONS FOR WOMEN'S SUBSTUDY
--Similar proportions of women and men in both arms achieved <50 c/ml among women, similar increases in CD4 counts were seen: significantly higher increases in CD4s were seen in women compared to men
--TDF arm showed significantly better total fasting lipid profile compared to men: lower increases from baseline in LDL & TG in women compared to men
--limb fat was higher in TDF arm compared to d4T arm
--mean decreases from baseline in BMD were higher in women on the TDF arm: no bone fractures were reported in women
--both arms showed similar renal safety
BACK TO OVERALL PUBLISHED STUDY IN JAMA
Resistance Analyses
Similar proportions of patients met the failure criteria for resistance analysis through week 144: 47 (16%) of 299 patients in the tenofovir DF group and 49 (16%) of 301 patients in the stavudine group (P = .91). Baseline resistance will be addressed in separate analyses. Postbaseline and baseline genotypic data were obtained for all patients with virologic failure. Mutations conferring resistance to efavirenz and lamivudine were observed most frequently.
The K65R mutation in HIV-1 reverse transcriptase can be selected by tenofovir DF and other NRTIs, and confers reduced antiviral activity to tenofovir, abacavir, didanosine, and lamivudine.28-32 K65R mutants retain activity to thymidine analogues (zidovudine and stavudine).31, 33-35 In this study, the K65R mutation occurred in 8 patients (7 prior to week 48, 1 from weeks 48-96, and none after week 96) administered tenofovir DF and 2 patients administered stavudine (P = .06). The K65R mutation was always accompanied by resistance to efavirenz or efavirenz plus lamivudine. Among patients in the tenofovir DF group who developed the K65R mutation, the median baseline HIV RNA level and CD4 cell count were 246 000 copies/mL and 24 cells/µL, respectively. In addition, the K65R mutation was associated with a mean 1.3-fold decrease in susceptibility to tenofovir DF (n = 8; range, 0.9-fold to 2.2-fold) without significant changes in susceptibility to zidovudine or stavudine. Reductions in susceptibility were observed for didanosine, abacavir, and lamivudine in vitro but only when the K65R mutation was present together with the lamivudine-associated M184V mutation (5 of 8 patients). One sample was reported as mixture of K65R/K (mutant and wild-type) along with M184V and had a fold-change of 0.9 fold (lowest of the group). Two samples exceeded the 1.4-fold cutoff for tenofovir in the PhenoSense assay (1.9- and 2.2-fold, one with M184V and one without). Overall, the M184V mutation appeared to be frequently responsible for improving the susceptibility to tenofovir in the presence of K65R, consistent with larger database analyses.33 Among the 8 patients who had developed the K65R mutation in the tenofovir DF group, 5 achieved an HIV RNA level of less than 50 copies/mL on their investigator-chosen second regimen with a median follow-up of 155 weeks, 2 patients were without follow-up, and 1 was nonadherent. The second-line regimens chosen for each patient were unique but all included a protease inhibitor and 1 to 2 other NRTIs. Two continued tenofovir DF in the subsequent regimen; both achieved complete virologic suppression.
|
|
NO OF MUTATIONS
|
|
|
|
TDF n=299
|
d4T n=301
|
|
|
Virologic Failure
|
47
|
49
|
|
|
Any EFV Resist mutat
|
26
|
24
|
|
|
--Alone
|
7
|
6
|
|
|
+M184V/I
|
10
|
12
|
|
|
+K65R
|
3
|
1
|
|
|
+K65R+M184V/I
|
5
|
1
|
|
|
+other NRTI resist
|
1
|
4
|
|
|
Any M184V/I
|
18
|
17
|
|
|
M184V/I alone
|
3
|
2
|
|
|
Any K65R mutat
|
8
|
2
|
p=.06
|
|
K65R alone
|
0
|
0
|
|
|
Wild-type or as baseline
|
18
|
23
|
|
INTRODUCTION
Highly active antiretroviral therapy has transformed human immunodeficiency virus (HIV) infection into a chronic manageable disease. However, although many regimens lower plasma viral load to below the limit of detection in most patients, maintaining a durable response remains challenging because of adverse effects, long-term toxicity, and complex dosing schedules, all of which can lead to nonadherence, virologic failure, and drug resistance.
Adverse effects and metabolic toxicity associated with protease inhibitor use have resulted in increasing use of regimens containing nonnucleoside reverse transcriptase inhibitors (NNRTIs) for initial therapy. However, some nucleoside analogue reverse transcriptase inhibitors (NRTIs) have also been associated with long-term toxicity, including lipoatrophy, lactic acidosis, and peripheral neuropathy.7 It has been proposed that these toxicities are caused by NRTI-induced damage to mitochondrial DNA.
Tenofovir disoproxil fumarate (tenofovir DF) is the first nucleotide analogue reverse transcriptase inhibitor approved for the treatment of HIV infection.9 It has a long intracellular half-life and is formulated as a single 300-mg tablet that is taken once daily. In vitro, tenofovir DF is a weak inhibitor of mitochondrial DNA polymerase gamma and appears not to affect the mitochondrial DNA content in multiple cell types. In a viral dynamics study of tenofovir DF monotherapy, antiretroviral-naive patients experienced a 1.6 log10 median decrease in HIV RNA over 3 weeks. The potency of tenofovir DF has been demonstrated in treatment-experienced patients. In placebo-controlled trials with treatment-experienced patients, the 24-week toxicity profile was similar to that of placebo, and in longer-term studies no significant toxicities have emerged after 96 weeks of follow-up.
The K65R mutation is selected by tenofovir DF in vitro and has been reported in treatment-naive and treatment-experienced patients.
Preliminary 96-week interim data on the efficacy and safety of tenofovir DF have been reported, as has its efficacy in the setting of coinfection with HIV-1 and hepatitis B virus. In antiretroviral-naive patients, the combination of tenofovir DF with lamivudine and efavirenz has been classified as a preferred regimen in the Department of Health and Human Services treatment guidelines.
To evaluate the safety and efficacy of tenofovir DF treatment in antiretroviral-naive patients, we conducted a randomized, double-blind trial comparing tenofovir DF with stavudine, both given in combination with lamivudine and efavirenz.
Study Population and Design
This study was conducted at 81 centers in South America, Europe, and the United States. Eligible adult patients were treatment-naive (no prior treatment with any NNRTI or protease inhibitor, <=4 weeks of prior treatment with NRTIs) and had plasma HIV RNA levels greater than 5000 copies/mL. These eligibility criteria were similar to those of another HIV clinical trial performed at that time.22 Patients were required to have adequate hematologic (absolute neutrophil count >=1000/µL, platelets >=50 x 103/µL, hemoglobin >=8.0 g/dL), hepatic (transaminases <=3 x upper limit of normal), and renal function (serum creatinine <1.5 mg/dL [<132.6 µmol/L] and calculated creatinine clearance [Cockroft-Gault formula] >=60 mL/min [>=1.00 mL/s]). There was no minimum CD4 cell count for study entry. There were no requirements for normal lipid profiles at entry into the study. Each participant provided written informed consent. An institutional review board or ethics committee reviewed and approved the study protocol and informed consent form for each study center.
Objectives
The primary objective of this study was to assess the equivalence of tenofovir DF vs stavudine in combination with lamivudine and efavirenz for the treatment of patients infected with HIV who were antiretroviral naive as determined by the proportion of patients in each regimen with plasma HIV RNA levels of less than 400 copies/mL. The secondary objectives of this study were to assess efficacy as measured by change in CD4 cell count and proportion of patients with HIV RNA levels of less than 50 copies/mL, and to compare the safety and tolerability of the 2 treatment regimens.
End Points
The primary efficacy end point was the proportion of patients with HIV RNA levels of less than 400 copies/mL at week 48. Patients with missing HIV RNA data and patients who added or switched antiretroviral medications were analyzed as having HIV RNA levels of more than 400 copies/mL. The secondary efficacy end points were the proportion of patients with HIV RNA levels of less than 50 copies/mL and the change in CD4 cell count from baseline at weeks 48, 96, and 144. Patients who discontinued blinded study medications were encouraged to remain in the study while off study medications. Safety was assessed by the frequency and severity of adverse events and clinical laboratory abnormalities.
Sample Size
The planned sample of 600 patients (300 per group) gave the study 80% power to establish equivalence between the 2 study groups. The calculations assumed a response rate of 75%24 in each treatment group.
The tenofovir DF--containing group was considered equivalent to the stavudine-containing group if the lower confidence bound for the difference between groups in the proportion with HIV RNA levels of less than 400 copies/mL was more than --10%. Antiretroviral treatment--naive studies using 3 drug regimens often use --10% to --13% as the lower bound of the equivalence criteria. A lower limit of --10% constitutes a more stringent and conservative equivalence criterion because a treatment difference of this magnitude is a small fraction of the additional benefit provided by the third drug in the regimen. There was no upper bound for the predefined CIs because the study was designed as a noninferiority trial (ie, equivalence) and the lower bound of the 2-sided 95% CI for the difference of the primary end point (the tenofovir DF group -- stavudine group) was compared with --10% to determine noninferiority of tenofovir DF relative to stavudine.
The secondary analyses of efficacy included the CD4 cell count change from baseline, mean change in HIV RNA from baseline, and the proportion of patients with HIV RNA levels of less than 50 copies/mL using both the ITT, M = F, antiretroviral Switch = F, and ITT, M = F analyses. The ITT, M = F was used in addition to ITT, antiretroviral Switch = F as a secondary end point to provide sensitivity analysis to further assess the robustness of the results. In addition, the ITT, M = F analysis is often used in other trials involving antiretroviral-naive patients. For calculating the mean change from baseline in HIV RNA and CD4 cell count, the ITT population with all available data was used. (Because patients were followed up in the study after permanent discontinuation of the study regimen, all available data collected after study regimen discontinuation were included [ITT, missing = excluded] in the analyses regarding changes from baseline in viral load and CD4 cell count). Forty-nine copies/mL was used for samples with viral load below the limit of quantification (<50 copies/mL).
RESULTS
A total of 602 participants were enrolled between June 9, 2000, and January 13, 2001. Two patients never received study drugs. The last enrolled patient completed all visits for the double-blind 144-week study on January 30, 2004. Baseline demographic and laboratory characteristics were comparable between the 2 groups: 75% men; 26% women; 64% white; 20% Black; 7% Hispanic. HIV RNA 4.91 log. Mean HIV RNA 81,300 copies/ml. 45% >100,000 c/ml. CD 280. CD4s <200: 39%; CD4 <50 15%.
Through 144 weeks, the study regimen permanent discontinuation rate was 82 (27%) of 299 in the tenofovir DF group and 100 (33%) of 301 in the stavudine group. Overall, there were 5 (2%) of 299 and 6 (2%) of 301 deaths in tenofovir DF and stavudine groups, respectively. Reported causes of death were pneumonia, respiratory failure, hepatic failure, sepsis, and Kaposi sarcoma; no death was assessed by the investigator as study drug-related.
Excluding patients with missing data and those who switched to nevirapine, 24 (8%) in the tenofovir DF group and 12 (4%) in the stavudine group added or switched an antiretroviral agent through week 48 (P = .04). Included in this group were 4 patients from the tenofovir DF group and 1 from the stavudine group who added zidovudine due to pregnancy and had HIV RNA levels of less than 50 copies/mL prior to pregnancy detection. In addition, 3 patients in the tenofovir DF group and none in the stavudine group had baseline resistance to efavirenz leading to treatment failure and a switch in therapy.
Through 144 weeks, data on subsequent antiretrovirals used following study regimen discontinuation were available for 41 (50%) of 82 patients in the tenofovir DF group and 46 (46%) of 100 in the stavudine group. The antiretroviral agents used (tenofovir DF vs stavudine groups) included zidovudine (16 vs 19), stavudine (14 vs 8), tenofovir DF (9 vs 15), abacavir (3 vs 6), didanosine (5 vs 4), and emtricitabine (1 vs 0). Tenofovir DF or stavudine were also used as a subsequent drug in patients who were originally randomized to those treatment groups since investigators were blinded to treatment assignment or patients may have discontinued due to non--tenofovir DF or non--stavudine-related toxicities such as efavirenz intolerance. Most patients continued on NNRTIs (22 tenofovir DF vs 25 stavudine) with fewer patients starting protease inhibitors (12 tenofovir DF vs 8 stavudine). The number of patients switching from efavirenz to nevirapine for efavirenz-associated neuropsychiatric toxicity was similar in both groups through week 144: 21 (7%) of 299 patients in the tenofovir DF group vs 26 (9%) of 301 patients in the stavudine group. There were 57 (19%) of 299 patients and 64 (21%) of 301 patients in the tenofovir DF and stavudine groups, respectively, with no viral load values at week 144. All but 3 of these patients also discontinued the study regimen prior to week 144. They were treated as failures or excluded in the ITT analysis.
In an ITT (missing = excluded [see Statistical Methods]) analysis, patients in both groups demonstrated a similar mean HIV RNA decrease from baseline (3.1 log10 copies/mL) at weeks 48 and 144. Through week 144, patients in the tenofovir DF and stavudine groups achieved a mean CD4 cell count increase of 263 and 283 cells/µL, respectively.
The calculated adherence rate using the rough estimate for adherence via the proxy approach based on the duration on study regimen was 98% for both the tenofovir DF and the stavudine group.
AUTHOR DISCUSSION
This is to our knowledge the first large 3-year, randomized, double-blind trial of antiretroviral therapy in treatment-naive patients. The primary end point (ITT, M = F, antiretroviral Switch = F) analysis accounts for not only missing patient data but also antiretroviral changes due to adverse effects, tolerability, or regimen potency. The primary end point was the achievement of an HIV RNA level of less than 400 copies/mL at week 48. By this analysis, the 95% CI slightly exceeded (by 0.4%) the lower predefined limit of --10%, narrowly missing the criteria for equivalence. However, equivalence was suggested in the secondary analyses using the more stringent measure of HIV RNA level of less than 50 copies/mL at week 48. Equivalence was also demonstrated at weeks 96 and 144. It is notable that the --10% lower predefined limit is more stringent than the --12% that has been reported in a similar randomized, double-blind antiretroviral-naive trial.25
The high proportion of patients achieving HIV RNA level below limit of quantification through 144 weeks in both regimens is presumed to be due to a combination of the potency of the drugs used and the tolerability and simplicity of the regimens. Because the 2 regimens were so effective, treatment failure was uncommon. The development of the K65R mutation was less common than resistance to efavirenz or lamivudine. This mutation appears to be the only pathway to tenofovir resistance among treatment-naive patients, analogous to observations in treatment-experienced patients.17-18 The K65R mutation was observed in 8 patients (1 patient after week 48) failing therapy in the tenofovir DF group through 144 weeks, which represents less than 3% of the total number of patients treated or 17% of those experiencing virologic failure in the tenofovir DF group. All viral isolates expressing the K65R mutation retained susceptibility to zidovudine and stavudine, and possibly to abacavir, didanosine, and tenofovir. As a single mutation, K65R was not associated with a sufficient decrease in susceptibility to result in phenotypic resistance. However, when combined with M184V, the fold-change in susceptibility typically exceeded the assay cutoffs for abacavir and didanosine. These findings are consistent with recent published data using this assay.33 Of the 8 patients who developed the K65R mutation, 5 achieved virologic success with subsequent second-line regimens (consisting of 2 unique NRTIs plus protease inhibitor) after a median follow-up of 155 weeks, demonstrating that second-line regimens can be effectively constructed after virologic failure with K65R and other regimen-associated mutations.
This trial demonstrated significantly more favorable lipid profiles in the tenofovir DF group and more patients required the addition of lipid-lowering agents in the stavudine group. The difference in lipid profiles between stavudine and tenofovir DF had not previously been well defined in a large double-blind study. Improvements in lipid abnormalities in patients switching from stavudine to tenofovir DF have recently been reported.38
The overall incidence of adverse events attributed to mitochondrial toxicity was significantly more among patients receiving stavudine. Significantly less total limb fat and a greater incidence of lipodystrophy were observed in the stavudine group. In the absence of effective treatments for fat loss,39-40 avoiding antiretrovirals associated with fat loss may be the best practice. Finally, patients in the tenofovir DF group continued to gain weight through 144 weeks, in contrast with the patients in the stavudine group who lost weight from week 24 to week 144. This initial weight gain through week 24 may arise from the initiation of antiretroviral therapy but the subsequent weight decline through 144 weeks may be a consequence of loss of peripheral limb fat.
Although decreased bone mineral density and nephrotoxicity have been observed in experiments with monkeys receiving high doses of subcutaneous tenofovir, in this 3-year trial decreases in bone mineral density were small and largely nonprogressive but significantly greater in the tenofovir DF group at the lumbar spine but not at the hip. There have been case reports of renal toxicity in patients with antiretroviral regimens containing tenofovir DF.41-43 However, through 3 years in this study in patients with normal renal function at baseline, the renal safety profile was similar between the tenofovir DF and stavudine groups.
Although lamivudine and stavudine were administered twice daily in this study, lamivudine is now approved for once-a-day administration. A stavudine extended-release tablet has been approved for once-a-day administration but is not currently available. Once-daily regimens are likely to be easier to adhere to and also allow for directly observed therapy in selected settings. Directly observed therapy has been shown to improve outcomes when used in the treatment of HIV infection.44
These data support the use of tenofovir DF as a component of initial therapy for HIV infection. They also provide further support for the use of efavirenz-based regimens in this patient population. Although both tenofovir DF and stavudine performed equally well with respect to antiviral potency, the 3-year results indicate that tenofovir DF was associated with less toxicity than stavudine.
|
| |
|
|
|
|
|