| |
Hepatitis C. Development of new drugs and clinical trials: Promises and pitfalls
|
| |
| |
Summary of an AASLD hepatitis single topic conference, Chicago, IL,
February 27-March 1, 2003
Hepatology
Volume 39, Issue 2, Pages 554-567
Jean-Michel Pawlotsky 1, John G. McHutchison 2 *1Department of Virology (EA 3489), Henri Mondor Hospital, University of Paris XII, Créteil, France2Duke Clinical Research Institute, Duke University Clinical Center, Durham, NC
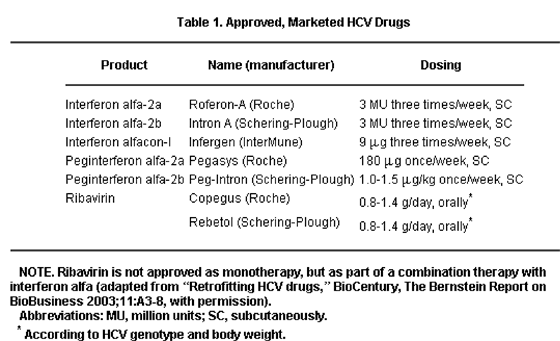
Chronic hepatitis C virus (HCV) infection affects a large number of patients worldwide. While our current therapies (Table 1) are effective in approximately 50% of patients, therapy is costly, prolonged, and associated with significant side effects, and it is not suitable for certain groups of patients. For these reasons, improved treatment regimens are necessary for patients with this disease. In February 2003, the annual American Association for the Study of Liver Diseases (AASLD) Single Topic Conference devoted to viral hepatitis and to fostering development of research in this field focused on recent advances in the process of antiviral drug design and new anti-HCV drugs currently in development. The drug design process, how these agents should be evaluated in future clinical trials, the potential problems and pitfalls related to pharmacodynamics and pharmacokinetics, viral resistance, clinical trial end points and design, and drug interactions, as well as HCV targets for novel drug development and the HCV drug pipeline were discussed. The preface and aims of the conference were to provide investigators with new knowledge and some of the necessary tools to aid in planning future rational clinical trial design in this rapidly changing field.
|
|
| |
| |
 |
|
| |
| |
Abbreviations
HCV, hepatitis C virus ; AASLD, American Association for the Study of Liver Diseases; PCR, polymerase chain reaction; IFN, interferon; NS3, nonstructural 3; IRES, internal ribosome entry segment; NC, noncoding; RdRp, HCV RNA-dependent RNA polymerase; DC-SIGN, dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin; L-SIGN, liver/lymph node-specific intercellular adhesion molecule 3-grabbing nonintegrin; LDL, low-density lipoprotein; TGF beta, transforming growth factor beta; PDGF, platelet derived growth factor; Th, T-helper; IL, interleukin; IMPDH, inosine monophosphate dehydrogenase; NK, natural killer; ALT, alanine aminotransferase; RNAi, RNA interference; siRNA, silencing RNA; BVDV, bovine viral diarrhea virus; HIV, human immunodeficiency virus; NNI, Nonnucleoside inhibitors; HCIg, hyperimmune anti-HCV immunoglobulin; ISCOMs, immunostimulating complexes; HGF, hepatocyte growth factor; PPAR, peroxisome proliferator activated nuclear receptors; FGF, fibroblast growth factor; TIMPs, tissue inhibitors of metalloproteinases.
PRE-CLINICAL STUDIES
New treatment for HCV requires the development of new agents with unique biological profiles that are effective against a broad range of viral genotypes. While drug or target discovery represents the beginning of the process, multiple additional studies are required before an antiviral agent becomes a clinically effective and safe drug. Using a modern systematic drug discovery and development process, the risk of failure in the clinic can be markedly reduced. In the case of small antiviral compounds, the process generally consists of the following steps (communication by R.F. Schinazi, Decatur, GA): - The first step is the screening of already existing libraries of potential compounds or the design and synthesis of new molecules based on potential target site structure knowledge.
- It is then necessary to rapidly test the potency and toxicity of potential agents - i.e. the selectivity of the targets and actions.
- The stability of the chemical entity and protein binding must be evaluated under various conditions.
- The mechanisms of action of the compound must be explored with appropriate in vitro tools.
- Toxicology studies must be run in at least two animal species. If the toxicity profile is acceptable, then the compound joins the hot list of compounds to proceed.
- The metabolism of the compound must be understood, and pharmacokinetics studies must be performed in animals.
- Efficacy studies must be performed in animals.
- The ultimate preclinical steps include various studies testing drug combinations in vitro and in vivo, selection of resistant viruses, viral fitness, pyrophosphorolysis, and others.
Efficacy
At the present time, the relative potency of anti-HCV agents currently can be determined rapidly and reproducibly by means of enzymatic and cellular systems coupled with quantitative real-time polymerase chain reaction (PCR). Efficacy models include tissue culture and animal models in which HCV replicates efficiently and antiviral molecules can be tested for their capacity to inhibit HCV replication.
Tissue Culture Systems.
Two models based on tissue culture appear to be of great interest for antiviral drug development. First, primary cultures of human hepatocytes isolated from uninfected patients, and infected in vitro with serum samples from HCV-infected patients represent the model closest to the physiologically infected cell.[1] They have been shown to be sensitive to infection and permissive to HCV replication.[1][2] This system has been used to show that interferon (IFN) alfa inhibits HCV replication in a dose-dependent manner.[2] It provides a very good model by which to study the antiviral effects of antiviral molecules in infected hepatocytes of human origin (communication by P. Maurel, Montpellier, France). Second, RNA replicons are subgenomic or genomic HCV RNA molecules that are capable of initiating and maintaining replication in the human Huh7 hepatoma line at relatively high copy numbers.[3-11] The subgenomic or genomic replicon systems provide excellent cell-based assays to evaluate inhibitors of HCV enzymes or nucleic-acid targeting strategies and are currently widely used for this purpose. Replicon-containing Huh7 cells have also been used to produce cell-free replication complex preparations that allow testing of inhibitors uncovered in screens using purified enzymatic components, and they can also be used to determine whether hits in cell-based replicon screens are capable of directly inhibiting RNA replication.[9-11] (communication by C.M. Rice, New York, NY).
Animal Models.
Animal models provide a crucial link between in vitro experimentation and the establishment of human clinical trials. The chimpanzee model has contributed to many advances in the HCV field,[12] but major impediments to the utilization of chimpanzees in medical research include the cost, availability, and ethical issues. In the tupaia, a tree shrew indigenous to Southeast Asia, HCV viremia is rare and, when it occurs, low and either transient or intermittent.[13] In the Trimera mouse model,[14] the practical window for drug administration is limited to a few days within the second week after transplantation, making this system difficult to utilize in drug screening experiments. Mercer's group recently humanized a murine liver through hepatocyte transplantation.[15] These mice with humanized livers are capable of de novo infection with HCV, generate viral RNA levels equivalent to those of humans (105 to 106 viral RNA copies/mL), may support stable viral production for weeks or months, and can transmit the virus to other noninfected murine recipients.[15] The efficacy of IFN alfa-2b, immunotherapies, antiviral gene therapy approaches, and pharmaceuticals that have shown promise in in vitro systems has been or is currently being tested in this model (unpublished data; communication by D.F. Mercer, Omaha, NE).
Pharmacology
The emphasis of pharmacological studies now has shifted to the early demonstration of pharmacological activity (proof of concept) or efficacy. This shift has been aided by the development of sophisticated methods of assessment, focusing on the use of biomarkers in decision-making.[16] At the preclinical stage, such models may assist initial human studies by identifying relevant exposure levels (communication by P. Glue, New York, NY).
Toxicity and Drug-Drug Interactions
There are two main types of safety risk: (1) mechanism-based toxicity, in which adverse effects are due to modulation of the same target or pathway that produces the desired therapeutic effect, and (2) compound-based toxicity, in which adverse effects are due to secondary actions of the drug candidate and are not related to the therapeutic mechanism. Both mechanisms must be addressed prior to further development by means of in vitro and in vivo preclinical toxicity screens. In addition, many drug-drug interactions can be correlated with the inhibition and-or induction of cytochrome P450 enzyme activity. Quantitative in vitro prediction of clinical interactions, however, remains difficult (communication by K.R. Romines, Research Triangle Park, NC).[17-19]
Clinical Trials
After the long and arduous preclinical process, a few survivor compounds may reach the list of lead candidate drugs for clinical evaluation. Clinical evaluation then generally includes four types of trials, from phase I to phase IV. Additional issues, including viral kinetics and resistance, must be addressed at these stages.
Viral Kinetics
Using viral load monitoring and mathematical models to analyze longitudinal data yields estimates of the antiviral efficacy of therapy. Several studies have described the kinetics of HCV during IFN alfa therapy in vivo.[20-25] Overall, these principles allow one to use viral kinetics to deduce features of the patient's response to therapy. They will prove to be particularly useful in evaluating the early efficacy of new HCV drugs, and understanding their mechanisms of action and eventual failure. They may also be important to predict the outcome of future therapy and derive appropriate clinical decision guidelines (communication by A.S. Perelson, Los Alamos, NM).[26]
Resistance.
Our current knowledge of HCV resistance to therapy includes partial understanding of the mechanisms underlying IFN alfa-based therapy failure to eradicate infection,[27] and a clearer view of the mechanisms underlying the resistance of other viruses (such as hepatitis B virus, human immunodeficiency virus, or herpes viruses) to specific inhibitor molecules. Viral resistance to specific antiviral drugs is characterized by the selection of minor viral populations bearing mutations conferring resistance to the specific drug. Treatment withdrawal is usually followed by restoration of the sensitive genotype and phenotype. Combined therapy with multiple drugs with different targets and mechanisms of action delays the emergence of resistant viruses, but multiresistant viruses may emerge after several years of treatment.[28] The natural variability of the HCV targets for new drugs, such as the nonstructural viral protein 3 (NS3) serine proteinase and internal ribosome entry segment (IRES),[29] suggests that resistant variants could be selected by future HCV inhibitors. Such events will need to be monitored with appropriate virological tools early in the clinical development of these drugs. If viral resistance becomes a clinical issue, multiple therapy combining drugs with different targets and modes of actions will probably become the standard of care (communication by J.M. Pawlotsky, Créteil, France).
Targets for New HCV Therapies
Targets for Antiviral Inhibitors
RNA structures and viral proteins playing a critical role in the HCV lifecycle represent ideal targets for specific HCV inhibitors.
5 and 3 Noncoding Regions.
The short 5 and 3 noncoding (NC) segments of the HCV genome contain structured RNA elements that are critically important for the initiation of protein translation, as well as for specific recognition of the termini of positive- and negative-strand RNAs by the HCV RNA-dependent RNA polymerase (RdRp) and subsequent initiation of RNA transcription.[30][31] Two general approaches to the inhibition of these key steps can be envisioned. First, molecules that cleave the 5 NC segment or otherwise destabilize its higher order structure will abrogate translation of the viral polyprotein. Such drug candidates may also disrupt initiation of plus-strand RNA synthesis, since the involved RNA signals are overlapping within the genome. Similar approaches could be adopted for the 3 NC segment, potentially eliminating the first step in viral RNA synthesis. Alternatively, the growing understanding of the structures of these RNA segments at atomic level resolution[31-33] should make it possible to rationally design small molecules that bind specifically to structures within the 5 NC and 3 NC segments and that could serve as inhibitors of translation, translation initiation, and/or RdRp recognition. In both cases, the target would be a relatively large RNA structure that most likely has multiple contacts with macromolecular protein assemblies (i.e., the 40S ribosome subunit or the replication complex[31][32]). It remains to be seen whether small molecules could effectively disrupt the resulting high affinity RNA-protein interaction (communication by S.M. Lemon, Galveston, TX).
p7.
Although its function is partly unknown, it has been suggested that the p7 protein could form ion channels in the endoplasmic reticulum membrane, and that this may be necessary to HCV replication. It therefore provides a potential target for antiviral intervention.
NS3 Serine Protease.
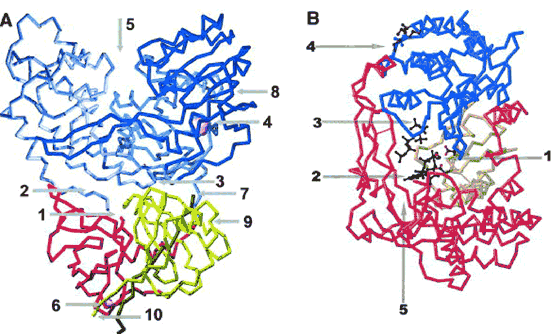
The NS3 serine protease belongs to the chymotrypsin class of proteases and is involved in HCV polyprotein maturation in which it cleaves an initially synthesized viral polyprotein into functional proteins. Three-dimensional (3-D) structures of the NS3 protease with and without the requisite viral cofactor NS4A were determined by x-ray crystallography.[34][35] Extended contacts are made between the polyprotein and NS3 protease during recognition of polyprotein processing sites. These contact regions potentially provide distinct types of inhibitor binding sites on NS3 (Fig. 1A).[36][37] Some NS3 protease inhibitors bind at subsites utilized by the polyprotein region on the N-terminal side of the scissile bond. Many of these inhibitors achieve increased affinity for the NS3 protease through additional interactions with the active site catalytic machinery including the active site serine and oxyanion hole. Inhibitors that span the active site and thus more closely mimic the polyprotein substrate have also been synthesized (communication by P.C. Weber, Yardley, PA).
|
|
| |
| |
 |
|
| |
| |
Figure 1. (A) Inhibitor sites on the NS3/4A protease/helicase. The backbone trace of the bifunctional NS3/4A is drawn from the crystallographic coordinates (1CU1). NS3 protease subdomains are shown in yellow and red, the central portion of the NS4A cofactor is shown in green, and the essential Zn is indicated by a sphere. The three helicase subdomains are shown in shades of blue. The NTPase subdomain is colored cyan and one of the NTP phosphate binding locations is indicated by a pink sphere. The arginine-rich subdomain that is structurally-homologous to the NTPase domain is colored dark blue, and the third helicase subdomain is colored light purple. Sites for competitive inhibitors include the protease active site (1), and here the serine and histidine side chains of the catalytic triad are shown. Arrows (2) and (3) indicate the polyprotein substrate P and P' recognition sites, respectively, and (4) and (5) indicate the NTPase- and ssRNA-binding sites of the helicase. Locations of additional RNA-recognition sites are unknown. Molecules that prevent productive binding of essential cofactors, such as the Zn atom (6) or NS4A cofactor peptide (7), also act as inhibitors. Inhibitors of requisite interdomain motions of the helicase subdomains bind near the end of the arrow (8). The general region of membrane attachment of the bifunctional protease/helicase is denoted by an arrow (9), and interference with attachment could also prevent formation of a competent replication complex. Finally, inhibitors preventing substrate recognition by the NS2/3 protease might bind near the NS3 N-terminus (10) prior to cleavage of the polyprotein (figure prepared by P.C. Weber). (B) Inhibitor sites on the NS5B polymerase. The backbone trace of the NS5B enzyme is drawn from the crystallographic coordinates of its binary complex with UTP (1GX6). The polymerase is shown in red, gold, and blue for the fingers, palm, and thumb subdomain, respectively. Sites for competitive inhibitors include the polymerase active site (1), and here the side chains of the catalytic aspartic acid residues are shown. Arrows (2) and (3) indicate the nucleotide binding sites for polymerization and for de novo initiation of RNA synthesis, respectively. Bound nucleotides are indicated in black. An arrow (4) indicates the location of the allosteric surface GTP binding site, a region at the connection between fingers and thumb where several nonnucleotide inhibitors have been shown to bind. In this region, the side chains of residues that contact the allosteric GTP are indicated in black (Pro 495 and 499, Arg 502). Finally, an arrow (5) shows the template RNA binding groove (communication by F.A. Rey).
NS3 Helicase.
The NS3 helicase primary function is to unwind the viral genomic RNA during replication. Studies probing the mechanism of polynucleotide unwinding have led to observations that may suggest new strategies for drug discovery.[38] In addition, an increasing number of 3-D helicase structure determinations, including a structure of the full-length NS3 protein having both protease and helicase domains[39] (Fig. 1A), has helped delineate the functionally important regions of the NS3 helicase. Structural comparisons revealed helicase domain motions that accompany the progressive unwinding of nucleic acid duplexes and that may serve as targets for specific inhibitors (Fig. 1A).[40] The NS3 helicase is characterized by the location of an NTP binding site near the interface between two structurally homologous nucleotide binding domains, the existence of a third domain composed primarily of alpha-helices, and the occurrence of a pronounced substrate binding groove, which binds an extended polynucleotide (communication by P.C. Weber).
NS5B RNA-Dependent RNA Polymerase.
The HCV RdRp (NS5B protein) is a 68 kDa protein that catalyzes RNA synthesis during replication. Several 3-D structures of active, water-soluble domains missing the hydrophobic C-terminal tail that localizes the RdRp to internal cellular membranes have been determined.[41-43] The HCV RdRp displays the classical Finger/Palm/Thumb motif observed in nucleic acid polymerases and a number of molecular interaction sites that can be targeted by specific inhibitors (Fig. 1B). It is, however, unique by its fully encircled active site into which nucleotides can bind in the absence of the template. The active site of the enzyme is a target for nucleoside/nucleotide analogue inhibitors. In addition, a specific GTP binding site has been identified at the surface of the enzyme, more than 30 Å away from the active site.[44] Nonnucleoside inhibitors of HCV RdRp were recently shown to bind very close to the surface GTP site, suggesting that a possible mode of action of these drugs is by inhibiting the conformational change that would be needed for RNA elongation.[45] Finally, indirect evidence suggests that some conformational changes must occur upon simultaneous binding of template and nucleotide to create the replication initiation platform. This mechanism could also serve as a target for antiviral drugs (communication by F.A. Rey, Gif-sur-Yvette, France).
Currently the most promising targets for new drug development appear to be the NS3 serine protease and RdRp (Fig. 1).
Alternative Targets
Other mechanisms of infection and disease may be suitable for future drug targets. The objectives may be to inhibit virus entry into cells by targeting the cellular receptor structures or their interactions with surface viral structures, to neutralize the virus or accelerate infected cell death by interacting with host immune responses, or to prevent liver disease progression by slowing or reversing fibrogenesis.
Virus Attachment and Entry Into Cells.
The HCV genome encodes two envelope glycoproteins, E1 and E2, which are thought to be expressed as noncovalent heterodimers at the virus surface. The mechanism by which HCV enters target cells is currently unknown. The E2 glycoprotein is thought to be responsible for initiating virus attachment, and the E1 glycoprotein has been hypothesized to contain the fusion peptide responsible for the fusion of the virus and cell membranes.[46] An early interaction of envelope glycoproteins with glycosaminoglycans has been suggested to play a role in cell recognition and tropism. Truncated soluble versions of E2 have been reported to bind specifically to human cells and were used to identify interactions with tetraspanin CD81,[47] with scavenger receptor class B type I,[48] and with dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) and liver/lymph node-specific intercellular adhesion molecule 3-grabbing nonintegrin (L-SIGN).[49] A role has also been suggested for the low-density lipoprotein (LDL) receptor.[50] The relevance of these molecules as receptors for HCV infection in vivo and as targets for future therapies is currently unknown and awaits the development of systems to study HCV-cell entry. One approach to overcoming the lack of a conventional cell culture system for HCV is to generate retroviral pseudotypes bearing HCV glycoproteins, which are infectious for a restricted number of human hepatoma cell lines.[51][52] This system will allow further understanding of the early events of HCV infection in detail,[51][52] to identify cellular molecules used by HCV, and to screen for small molecules able to block viral entry (communication by J.A. McKeating, New York, NY). This system will also be useful to assess neutralizing responses in vitro and to test therapies based on virus neutralization, such as specific anti-HCV immunoglobulins.
Immune Responses.
The cellular immune response plays a major role in HCV infection and could represent an attractive target for therapeutic intervention. Indeed, vigorous and multispecific CD4+ and CD8+ T-cell responses during acute hepatitis C are associated with viral clearance and recovery.[53] Moreover, HCV-specific memory T cell responses remain detectable in the peripheral blood for decades[54] and mediate rapid viral clearance upon reexposure to the virus.[55][56] In contrast, a lack of this response or a failure to maintain it for a sufficient time, particularly in the face of viral mutations, is associated with development of persistent infection and chronic hepatitis.[57] Once persistent hepatitis is established, the frequency of HCV-specific T cells is typically low, and their proliferative, cytokine, and cytotoxic effector functions appear to be impaired.[58] Whether successful antiviral therapy leads to transient and/or long-lasting reconstitution of cellular immune responses is controversial.[59] With regard to the future role of immunotherapy, our current knowledge of HCV-specific cellular immune responses raises the following questions: Should antiviral therapy be combined with approaches to modulate and/or induce the cellular immune response in order to induce long-lasting immunity, and does immunomodulatory therapy accelerate infected cell clearance and improve sustained virological response rates in chronic hepatitis C? (communication by B. Rehermann, Bethesda, MD).
Fibrosis Progression.
If the virus cannot be eradicated, an alternative approach would be to slow the progression of liver disease. Hepatic fibrosis is the major histologic complication of chronic HCV infection, and it generally develops after many years, leading to cirrhosis within an estimated 10-50 years. A key pathogenic event in fibrosis progression is the transition of hepatic stellate cells from a quiescent to an activated state. This process is characterized by the production of increased amounts of extracellular matrix and de novo expression of smooth muscle alfa-actin, the latter characteristic consistent with cellular transformation to myofibroblasts.[60-62] One of the most remarkable aspects of this response is enhanced extracellular matrix production, or fibrogenesis. Fibrogenesis after injury to the liver is characterized by significant increases in collagen (type I>III>IV) and other extracellular matrix constituents such as laminin, fibronectin, and proteoglycans (dermatan sulfate, chondroitin sulfate, heparan sulfate).[61][63][64] This wounding process is complex and integrated, and it involves aspects of matrix synthesis and deposition, as well as degradation. Cross-talk between stellate cells and the extracellular matrix appears to play a critical role in fibrogenesis, as well as a number of cytokines and small peptides, including transforming growth factor (TGF) beta, platelet derived growth factor (PDGF), endothelin, and angiotensin II. New specific, antifibrotic therapies may target at reducing or removing the primary injury, inhibiting stellate cell activation, stimulating matrix degradation, stimulating stellate cell apoptosis, or blocking the downstream effects of stellate cell activation (communication by D.C. Rockey, Durham, NC).
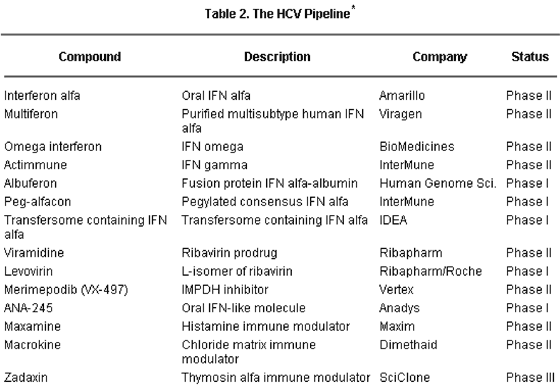
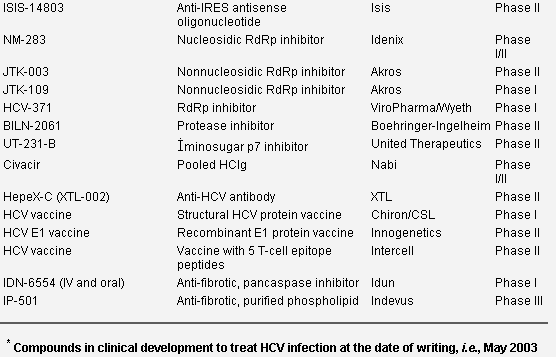
New HCV Drugs: The Pipeline
Various new directions are currently being explored in HCV drug development, and several specific and nonspecific anti-HCV drugs have already reached the clinical development phase (Table 2).
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
New Directions in IFN-Ribavirin-Based Therapy
Human IFNs have been classified according to the cell surface receptor with which they bind. Type 1 IFNs bind to the IFN alpha heterodimeric receptor IFNAR1/IFNAR2 and include the 21 nonallelic subtypes IFN alfa, IFN beta, IFN omega, and IFN tau.[65] IFN gamma binds to a unique cell surface receptor, is classified as a type 2 IFN, and displays antiviral, antifibrotic, and immunomodulating activity that effects the T-helper (Th) 1 response.[66] Recently, three novel cytokines have been identified that share sequence identity with type 1 IFNs and bind to a novel cell surface receptor. These putative type 3 IFN molecules have been called interleukin (IL) 28A, IL-28B and IL-29. They are induced by viral infections and exhibit marked antiviral activity in vitro.[67]
New IFNs.
Challenges still exist with respect to IFN treatment optimization, including poor pharmacokinetic profiles, limited biological activity, suboptimal therapeutic indices, and limited knowledge on non-alfa, non-beta IFNs efficacy. Several groups have attempted to modify naturally occurring IFNs to improve their performance. These modifications include alteration of the primary amino acid sequences, the addition of polyethylene glycol, alterations of glycosylation patterns, and the production of fusion proteins. Consensus IFN alfa (IFN-alfacon1) is a second-generation cytokine that was engineered to contain the most frequently occurring amino acids among the nonallelic IFN alfa subtypes. IFN alfacon1 has been shown to be more effective than naturally occurring type 1 IFNs in cell culture models and equally effective in clinical trials.[68][69] Other novel alfa IFNs have been produced by gene-shuffling the family of 20 human IFN alfa DNA encoding sequences.[70] Using this technique, a novel nonnaturally occurring type 1 IFN has been described with a 285,000-fold increase in antiviral activity when compared with IFN alfa-2b. The role of this highly active IFN in the treatment of chronic hepatitis C has yet to be elucidated.
The addition of polyethylene glycol to therapeutic IFN alfa proteins has been shown to dramatically increase plasma exposure following dosing and has led to increased response rates.[71][72] Recently, an IFN beta molecule has been conjugated to a linear 20-kDa polyethylene glycol molecule targeted at a single site on the N-terminal amine.[73] This molecule is now entering clinical trials for the treatment of chronic hepatitis C. A pegylated form of IFN-alfacon1 is also being clinically evaluated. Finally, a fusion protein of IFN alfa-2 and human serum albumin, has entered clinical trials for the treatment of chronic hepatitis C.[74] This molecule displays similar in vitro antiviral and antiproliferative activity to unmodified IFN alfa-2, but has marked improvements in pharmacokinetic characteristics. Several new techniques for selection of novel second-generation IFNs have produced even more potent molecules, and clinical trials of these molecules are either underway or planned for the near future. Lastly, the role of additional naturally occurring IFN species, such as IFN gamma and IFN omega, is now being elucidated in clinical trials (communication by L.M. Blatt, Brisbane, CA).
Oral IFN Inducers.
Oral IFN inducers represent a class of compounds that may allow the generation of an effective immune response by induction/modulation of cytokine responses at the site of infection, or that may supplement or replace parenteral administration of IFN. The central challenge in the use of such agents as oral therapy for chronic HCV infection is the delivery of effective doses to the liver. Agents known to induce IFN alfa and other cytokines include (among many) relatively high molecular-weight agents, such as double-stranded RNA (poly I:C), and CpG oligonucleotide derivatives. In contrast to the complexities associated with oral delivery of such large molecules, low-molecular-weight molecules may offer similarly useful immune-modulating properties and a reasonable probability of oral absorption. Two advanced chemical classes include the imidazoquinolones imiquimod and resiquimod,[75] and the nucleoside analogs ANA245 and ANA971.
Imiquimod is approved for use as a topical agent in dermatology. Toxicity probably resulting from cytokine induction was reported in human studies.[76][77] Resiquimod is currently in phase II studies for chronic viral hepatitis, and preliminary reports indicate a lack of specific detectable antiviral activity (Pockros et al., poster presented at the meeting). ANA245 is a low-molecular-weight nucleoside analog that induces multiple cytokines, including IFN alfa, and it showed immunologically mediated antiviral activity in a range of viral infection models (D.R. Averett, unpublished data, May 2003). However, its oral bioavailability is limited at high doses. Studies in humans are underway to characterize the safety and pharmacokinetics of ANA245 after intravenous administration, as a component of the development program for ANA971, a novel molecule that was shown to efficiently deliver ANA245 at levels associated with antiviral effects to the plasma of various animals (Averett et al., unpublished data) (communication by D.R. Averett, San Diego, CA).
Ribavirin-Like Molecules.
Ribavirin monotherapy is ineffective in inducing sustained viral clearance, but it significantly enhances the sustained viral clearance rate when combined with IFN. Until now, the mechanism by which ribavirin enhances IFN efficacy remains unknown. There are four proposed mechanisms of action: immune-mediated activity on the host Th1/Th2 balance; inhibition of host enzyme inosine monophosphate dehydrogenase (IMPDH) activity; weak inhibitory activity against the RdRp; and induction of RNA mutagenesis.[78] Hemolytic anemia is a frequent side effect that limits ribavirin dosing, emphasizing the need for alternative molecules with similar mechanisms and efficacy and less toxicity.
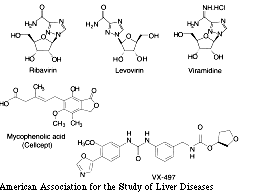
Levovirin is the L-sugar analogue of ribavirin (Fig. 2), with similar Th1/Th2 immunomodulatory activity. However, as an L-isomer, it does not undergo metabolism to the phosphate metabolites and hence, it does not inhibit the host IMPDH nor accumulate in erythrocytes, the mechanism responsible for hemolytic anemia.[79] In preclinical studies, levovirin was well tolerated by animals and did not have mutagenic effects in conventional short-term in vitro and in vivo assays. Phase I studies in humans showed that the drug is orally absorbed and well tolerated. A proof-of-concept study in combination with pegylated IFN alfa is under way. Viramidine is the amidine version of ribavirin (Fig. 2) that can be converted by adenosine deaminase to ribavirin.[80] The first pass effect of oral dosing will lead to the delivery of viramidine to the liver. As the liver is rich in deaminases, viramidine is converted to ribavirin and its phosphorylated metabolites and may be preferentially retained in the liver, establishing an equilibrium in which more viramidine/ribavirin are concentrated in the liver compared with other tissues, including erythrocytes.[79] Rodent and chimpanzee experiments confirmed that viramidine does target the liver, and early studies suggest acceptable safety, pharmacology, and toxicology profiles. In phase I studies, viramidine showed a similar profile of adverse events similar to those of ribavirin. However, the extent of hemoglobin drop with the highest dose was lower than the hemoglobin drop with conventional combination therapy. A phase II proof-of-concept study in combination with pegylated IFN alpha is ongoing. IMPDH inhibitors, such as mycophenolic acid (Cellcept) and VX-497,[81] are also currently being studied in patients with chronic hepatitis C (Fig. 2), but preliminary results showed no/minimal direct antiviral efficacy, similar to ribavirin (communication by J.Y.N. Lau, Newport Beach, CA).
|
|
| |
| |
 |
|
| |
| |
Figure 2. Chemical structure of ribavirin, levovirin (an L-sugar analogue of ribavirin), viramidine (the amidine version of ribavirin), and two IMPDH inhibitors, mycophenolic acid and VX-497 (communication by J.Y.N. Lau).
Other Immunomodulatory Drugs.
Several parenterally administered immunomodulatory drugs are currently being used in combination with IFN or pegylated IFN in clinical trials. They include: (1) histamine dihydrochloride,[82] which inhibits phagocyte-derived oxidative stress and inflammation and is currently being studied in phase II trials in combination with pegylated IFN and ribavirin, and (2) thymosin alfa-1, which promotes T-cell maturation and natural killer (NK) cells and differentiation of pluripotent stem cells. Preliminary results with this drug and IFN alfa were unclear, and thymosin alfa-1 is currently being evaluated in large phase II/III trials in combination with pegylated IFN alfa[83] (communication by D.R. Averett). Finally, IL-10, an anti-inflammatory drug, failed to show any beneficial histologic and antifibrotic effects in a randomized controlled trial, whereas a phase II study of IL-12, a different proinflammatory drug, suggested a lack of efficacy and significant toxicity.[84][85]
Specific Antiviral Inhibitor Molecules
Specific antiviral molecules target functional viral components, including viral RNA structures and viral enzymes.
Nucleic Acid-Based Therapy: Antisense Oligonucleotides, Ribozymes, and siRNAs.
The aim of the antisense approach is to exploit the high affinity and selectivity of nucleic acid hybridization for the development of highly specific drugs. Viral genomes contain numerous unique nucleic acid sequences not present in the human genome that have the potential to act as a virus-specific antisense target. Several groups have identified antisense oligonucleotides that inhibit the translation of HCV RNA in cell-free systems and cell culture models.[86][87] Among them, ISIS 14803, a 20-nucleotide antisense oligodeoxynucleotide that is complementary to the IRES region surrounding the translation initiation codon has entered clinical development. ISIS 14803 decreases HCV RNA and protein levels in various in vitro and in vivo models. The HCV RNA decrease was shown to occur through RNase H cleavage of the RNA in oligonucleotide-RNA heteroduplexed regions, and potentiated the antiviral effect related to inhibition of polyprotein translation. Two clinical studies of ISIS 14803 monotherapy have been conducted.[88][89] Reductions in plasma HCV RNA levels have been observed in 3 of 10 patients treated at 2 mg/kg (-1.3 to -2.2 log10) and in 6 of 20 patients dosed twice weekly at 6 mg/kg (-1.0 to -3.8 log10). Transient asymptomatic alanine aminotransferase (ALT) flares, up to 30 times, were observed in temporal association with plasma HCV RNA reductions and in other patients with no HCV RNA reductions.[88][89] The mechanism by which HCV RNA reductions occurred is unclear (communication by T.J. Kwoh, Carlsbad, CA).
Ribozymes, another nucleic acid-based strategy, are catalytic RNA molecules that cleave specific RNA sequences. They contain a catalytic core region flanked by binding arms with specific nucleotide sequences determined by the complementary base sequence of the target RNA. Heptazyme is a synthetic, stabilized 33-mer ribozyme that is chemically modified for resistance to enzymatic and chemical degradation. This first-generation ribozyme product was developed to treat chronic hepatitis C. Preclinical results indicated that Heptazyme selectively cuts hepatitis C RNA, significantly inhibiting viral replication in cell culture.[90] Phase II results indicated that Heptazyme as monotherapy led to a reduction in serum HCV RNA levels in 10% of patients. The development of Heptazyme has been halted in favor of developing a next-generation product with an improved therapeutic index. This strategy capitalizes on recent technological advances resulting in improved stability and targeting of ribozymes to specific tissues (communication by N. Usman, Boulder, CO).
A related and new potential class of therapeutics makes use of RNA interference (RNAi), a recently discovered process whereby cells down-regulate gene expression through destruction of a specifically targeted mRNA.[91] The RNAi process is mediated inside the cell by a naturally occurring protein complex that uses double-stranded RNA as a molecular guide to down-regulate genes at the posttranslational level. Rational design of double-stranded RNA permits the down-regulation of virtually any gene. In human cells, these double-stranded RNAs, known as silencing RNAs (siRNAs), show biological activity as short fragments of 20-23 residues. Stabilized siRNA compounds are currently being evaluated in the preclinical setting for their potential activity against HCV (communication by N. Usman).
p7 Inhibitors.
Long-alkyl-chain iminosugar derivatives, which are known to have antiviral activity against bovine viral diarrhea virus (BVDV),[92] were recently shown to inhibit HCV p7 ion channels,[93] suggesting these compounds with low toxicity profiles in animals might be used in the treatment of chronic hepatitis C. One such compound is currently in a phase II clinical trial.
HCV Protease Inhibitors.
The search for inhibitors of the NS2/NS3 cleavage reaction has been hampered by the hydrophobic nature of the protein and by the autocatalytic nature of the cleavage. In contrast, a number of peptide-based or peptidomimetic inhibitors have been developed for the NS3 serine protease and tested in vitro. Most of these inhibitors fall in one of three classes: (1) substrate analogues, (2) serine-trap inhibitors (or transition-state analogues), and (3) product analogues. In addition, efforts to discover nonpeptide inhibitors have also been made.[94] Optimization of an N-terminal hexapeptide cleavage product of a dodecapeptide substrate led to the discovery of BILN 2061, a small, selective, and potent inhibitor of the NS3 serine protease.[95] Inhibitor constant values of 0.30 mmol/L and 0.66 mmol/L with a noncovalent, competitive mode of inhibition were obtained for genotypes 1a and 1b, respectively. BILN 2061 was shown to retain its inhibitory efficacy in human cells and showed low nanomolar inhibition of HCV RNA replication through blockade of the NS3 protease-dependent polyprotein processing in the replicon system. BILN 2061 was administered for 48 hours in HCV-infected patients in early clinical development studies. Administration resulted in a rapid, dose-dependent HCV RNA decrease up to -4 log10 within 2 days at the highest doses, with a progressive return to baseline levels within a week after treatment withdrawal[96][97] (communication by D. Lamarre, Laval, Québec, Canada). Another NS3 serine protease inhibitor, VX-950, will soon enter clinical development.
HCV NS3 Helicase Inhibitors.
A few small-molecule inhibitors of the NS3 helicase with activity in vitro have been reported, but their inhibitory mechanisms, specificity, and potential efficacy in the clinical setting remain unclear.[94]
HCV RdRp Inhibitors.
Inhibitors of viral polymerases can be classified into three categories. (1) Nucleoside (substrate) analogues (cyclic or acyclic) are usually phosphorylated to their corresponding nucleoside triphosphate (nucleotide) in the cytoplasm of infected cells. The nucleotide is then typically incorporated by the viral polymerase during processive nucleic acid synthesis, leading to early termination and thus inhibition of the virus life cycle. Nucleoside inhibitors of viral polymerases are used therapeutically for human immunodeficiency virus (HIV), hepatitis B, and herpes viruses. (2) Nonnucleoside inhibitors (NNI) of clinical interest have thus far been described only for the HIV-1 reverse transcriptase. These compounds bind to an allosteric site on the enzyme surface away from the enzyme active site, possibly distorting the precise geometry of the enzyme active site so the enzymatic function is suppressed. (3) Foscarnet (phosphonoformic acid) is the prototypic and the only approved member of the pyrophosphate (product) analogue class. It is used in the treatment of infection by the herpes virus family. These agents are thought to interact directly with the pyrophosphate-binding site of the viral polymerases. Inhibitors of the HCV RdRp that belong to each of the above classes have been identified through the use of high throughput screening and rational drug design.[98] Several of these compounds have encouraging preclinical profiles, including the capability to inhibit HCV RNA replication in cell culture. Preliminary in vitro results suggest that resistance to RdRp inhibitors may occur in the clinical setting (communication by R. de Francesco, Pomezia, Italy).
A few orally bioavailable inhibitors of the HCV RdRp are under study in early clinical trials, such as JTK-003 and JTK-109, the potential mechanisms of which remain unclear, or NM283. NM283 is an orally bioavailable prodrug of a ribonucleoside analogue, NM107, that was discovered through its potent activity against BVDV in cell culture. NM107 triphosphate is a competitive inhibitor of purified BVDV polymerase in vitro (Ki 160 nM) and acts as a chain terminator of BVDV RNA synthesis. The efficacy of NM283 against HCV genotype 1 was established in a 1-week oral dosing study in chronically infected chimpanzees. Median drops in HCV viral load of -0.83 and -1.05 log10 were observed at the end of the dosing period in two animals per group in response to 8.3 and 16.6 mg/kg/day of NM283, respectively, compared with a placebo control animal. NM283 recently entered early phase clinical trials (communication by R.F. Schinazi).
Immune Therapy
Modulation of the host immune response to infection may include enhancement and broadening of the cellular immune response against HCV, inhibitory or immunostimulatory cytokine therapy, passive transfer of hyperimmune anti-HCV immunoglobulins (HCIg), and application of therapeutic vaccines.
Hyperimmune Anti-HCV Immunoglobulins.
Experimental studies with chimpanzees provided compelling evidence that the neutralization of epitopes located in the hypervariable region 1 of the HCV envelope gene prevented infection of susceptible animals.[99][100] It has been hypothesized that multiple infusions of HCIg containing neutralizing antiviral antibodies may modify virus replication and the clinical course of the infection. Therapeutic HCIg could also be particularly valuable in preventing recurrent hepatitis C in HCV-infected liver transplant recipients. HCIg has been prepared from virus-inactivated, HCV RNA-negative, 5% IgG obtained from 460 anti-HCV-positive plasma donors. In three chronically experimentally infected chimpanzees, passive HCIg transfer decreased the HCV RNA load in two chimpanzees and decreased ALT levels in all three animals once the level of passively transferred anti-HCV E2 reached a plateau. Both markers returned to baseline when HCIg infusions were stopped.[101] In three other chimpanzees acutely infected with HCV, multiple infusions of HCIg significantly shortened the length of HCV viremia and prevented acute hepatitis in comparison with control animals treated with immunoglobulin preparations without anti-HCV. HCV RNA disappeared from serum after a significantly shortened period of viremia in two animals, but it recurred in one of the two, when the level of anti-HCV E2 declined to negative values. The third animal had a fluctuating pattern of serum HCV RNA levels closely related to the changing level of anti-HCV E2 resulting from HCIg infusions. Recurrent HCV viremia was followed by enzymatic and histopathologic evidence of either acute or chronic hepatitis.[101][102] The mechanisms by which HCIg affected the rate of HCV replication require further study. A clinical trial is in progress in infected patients to investigate the safety and pharmacokinetics of HCIg in liver transplant recipients (communication by K. Krawczynski, Atlanta, GA).
Therapeutic Vaccines.
Recent data on immune responses in the chronic phase of infection suggest that a therapeutic vaccine capable of stimulating functional CD4+ and CD8+ T-cell responses in the chronic carrier may be of benefit.[58][103] To this end, various HCV recombinant polypeptide and plasmid DNA vaccines have been tested in nonhuman primates and shown to be capable of priming broad, functional CD4+ and CD8+ T-cell responses. Figure 3 shows various possible approaches to therapeutic vaccination. A recombinant E1/E2 vaccine was shown to prime the induction of viral neutralizing antibodies and CD4+ T-cell responses, and it prevented infection or evolution toward chronicity in a significant number of chimpanzees tested relative to control animals. Another vaccine formulation, based on yeast-derived fusion polyprotein comprising HCV genotype 1 NS3-NS4-NS5-core sequences combined with an immunostimulating complexes (ISCOMs) adjuvant, was capable of priming broad CD4+ and CD8+ T-cell responses in chimpanzees. It is now intended to test optimal formulations in chronic hepatitis C patients for their ability to increase the long-term response to pegylated IFN plus ribavirin (communication by M. Houghton, Emeryville, CA).
Other experiments have shown that cellular immune responses to the E1 envelope protein are almost absent in patients with chronic active hepatitis C, while long-term responders to IFN alfa therapy have, on average, higher levels of E1 antibodies.[104][105] A clinical-grade HCV E1 protein produced and purified from mammalian cells is in use for phase I and II clinical trials of therapeutic vaccination. In a healthy volunteer study, the E1/alum vaccine successfully stimulated both humoral and cellular immune responses. Signs and symptoms of inflammation at the site of injection were present in a minority of the volunteers and were the only reported adverse events.[106] In a subsequent phase IIa study, conversion from a negative to a strong E1-specific Th response was observed in the vast majority of patients. The levels of anti-E1 antibodies increased on average 3- to 4-fold after the second course of E1 injections. The proportion of patients showing a significant T-cell response to E1 increased from 9% at baseline to 91% at study end. The T-cell responses included a strong Th1 component with high IFN gamma levels. The E1-treated patients showed a significant decline of ALT as compared with baseline, and liver fibrosis had improved by one point or more in 38% of the patients (both correlated with the increase in E1 antibody levels). HCV E2 antigen levels in the liver declined, while serum HCV RNA levels remained unchanged. Further patient follow-up and maintenance therapeutic vaccinations are being conducted to examine the long-term disease-modifying effects, and additional placebo-controlled studies are ongoing (communication by G. Maertens, Gent, Belgium).
Antifibrotic Approaches
The most effective way to eliminate hepatic fibrosis is to clear the primary cause of liver disease. Therefore, curative antiviral therapy remains the best antifibrotic approach. However, when this goal cannot be achieved, several antifibrotic approaches are theoretically possible, for which various agents are currently being evaluated (communication by S.L. Friedman, New York, NY):
--Reduce inflammation or the host response to avoid stimulating stellate cell activation
--Directly down-regulate stellate cell activation
--Neutralize proliferative, fibrogenic, contractile and- or proinflammatory responses of stellate cells
--Stimulate apoptosis of stellate cells
--Increase the degradation of scar matrix.
Conclusion
The clinical research environment and the rapid tempo related to new drug development for patients with hepatitis C is exciting. While many opportunities exist to improve the outcome for our patients with newer and more targeted therapies, the process will be arduous, academically stimulating, associated with many promises and pitfalls, but eventually rewarding. As outlined in this conference, and as investigators and practitioners, we must consider and incorporate numerous preclinical, clinical, and regulatory issues into the conduct of our clinical trials related to this disease. The aim will be to achieve patient-centered, high-quality clinical research with meaningful results that we can convey to our patients without reservation. A large number of HCV functions and disease features can now be targeted for drug development. As a result, many new HCV drugs are currently in the pipeline, with several of them having already reached the clinical phase of development. It is clear that only a few of these candidates will finally be approved for clinical use in the treatment of HCV infection. It is, however, encouraging to foresee multiple therapies with complementary targets that could lead to cure of infection in a greater proportion of patients.
REFERENCES
- Fournier C, Sureau C, Coste J, Ducos J, Pageaux G, Larrey D, Domergue J, et al. In vitro infection of adult normal human hepatocytes in primary culture by hepatitis C virus. J Gen Virol 1998; 79: 2367-2374. Links
- Castet V, Fournier C, Soulier A, Brillet R, Coste J, Larrey D, Dhumeaux D, et al. Alpha interferon inhibits hepatitis C virus replication in primary human hepatocytes infected in vitro. J Virol 2002; 76: 8189-8199. Links
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999; 285: 110-113. Links
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 2002; 76: 13001-13014. Links
- Guo JT, Bichko VV, Seeger C. Effect of alpha interferon on the hepatitis C virus replicon. J Virol 2001; 75: 8516-8523. Links
- Krieger N, Lohmann V, Bartenschlager R. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol 2001; 75: 4614-4624. Links
- Lohmann V, Korner F, Dobierzewska A, Bartenschlager R. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J Virol 2001; 75: 1437-1449. Links
- Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 2003; 77: 3007-3019. Links
- Ali N, Tardif KD, Siddiqui A. Cell-free replication of the hepatitis C virus subgenomic replicon. J Virol 2002; 76: 12001-12007. Links
- Hardy RW, Marcotrigiano J, Blight KJ, Majors JE, Rice CM. Hepatitis C virus RNA synthesis in a cell-free system isolated from replicon-containing hepatoma cells. J Virol 2003; 77: 2029-2037. Links
- Lai VC, Dempsey S, Lau JY, Hong Z, Zhong W. In vitro RNA replication directed by replicase complexes isolated from the subgenomic replicon cells of hepatitis C virus. J Virol 2003; 77: 2295-2300. Links
- Lanford RE, Bigger C. Advances in model systems for hepatitis C virus research. Virology 2002; 293: 1-9. Links
- Xie ZC, Riezu-Boj JI, Lasarte JJ, Guillen J, Su JH, Civeira MP, Prieto J. Transmission of hepatitis C virus infection to tree shrews. Virology 1998; 244: 513-520. Links
- Ilan E, Arazi J, Nussbaum O, Zauberman A, Eren R, Lubin I, Neville L, et al. The hepatitis C virus (HCV)-Trimera mouse: a model for evaluation of agents against HCV. J Infect Dis 2002; 185: 153-161. Links
- Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, et al. Hepatitis C virus replication in mice with chimeric human livers. Nat Med 2001; 7: 927-933. Links
- Lesko LJ, Rowland M, Peck CC, Blaschke TF. Optimizing the science of drug development: opportunities for better candidate selection and accelerated evaluation in humans. Pharm Res 2000; 17: 1335-1344. Links
- Nebert DW, Russell DW. Clinical importance of the cytochrome P450. Lancet 2002; 360: 1155-1162. Links
- Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov 2002; 1: 259-266. Links
- Wienkers LC. Factors confounding the successful extrapolation of in vitro CYP3A inhibition information to the in vivo condition. Eur J Pharm Sci 2002; 15: 239-242. Links
- Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 1998; 282: 103-107. Links
- Neumann AU, Lam NP, Dahari H, Davidian M, Wiley TE, Mika BP, Perelson AS, et al. Differences in viral dynamics between genotypes 1 and 2 of hepatitis C virus. J Infect Dis 2000; 182: 28-35. Links
- Zeuzem S, Herrmann E, Lee JH, Fricke J, Neumann AU, Modi M, Colucci G, et al. Viral kinetics in patients with chronic hepatitis C treated with standard or peginterferon alpha2a. Gastroenterology 2001; 120: 1438-1447. Links
- Layden JE, Layden TJ, Reddy KR, Levy-Drummer RS, Poulakos J, Neumann AU. First phase viral kinetic parameters as predictors of treatment response and their influence on the second phase viral decline. J Viral Hepat 2002; 9: 340-345. Links
- Bekkering FC, Brouwer JT, Hansen BE, Schalm SW. Hepatitis C viral kinetics in difficult to treat patients receiving high dose interferon and ribavirin. J Hepatol 2001; 34: 435-440. Links
- Bekkering FC, Brouwer JT, Leroux-Roels G, Van Vlierberghe H, Elewaut A, Schalm SW. Ultrarapid hepatitis C virus clearance by daily high-dose interferon in non-responders to standard therapy. J Hepatol 1998; 28: 960-964. Links
- Bekkering FC, Stalgis C, McHutchison JG, Brouwer JT, Perelson AS. Estimation of early hepatitis C viral clearance in patients receiving daily interferon and ribavirin therapy using a mathematical model. HEPATOLOGY 2001; 33: 419-423. Links
- Pawlotsky JM. Antiviral treatment efficacy and failure in chronic hepatitis C. Antiviral Res 2003; 59: 1-11. Links
- Hirsch MS, Brun-Vezinet F, D'Aquila RT, Hammer SM, Johnson VA, Kuritzkes DR, Loveday C, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society-USA Panel. JAMA 2000; 283: 2417-2426. Links
- Soler M, Penin F, Ray S, Dhumeaux D, Pawlotsky JM. Hepatitis C virus NS3 serine protease variability and evolution are strongly constrained by the need for structural and functional conservation. J Hepatol 2002; 36( suppl 1): 5. Links
- Rijnbrand R, Bredenbeek PJ, Haasnoot PC, Kieft JS, Spaan WJ, Lemon SM. The influence of downstream protein-coding sequence on internal ribosome entry on hepatitis C virus and other flavivirus RNAs. RNA 2001; 7: 585-597. Links
- Yi M, Lemon SM. Structure-function analysis of the 3 stem-loop of hepatitis C virus genomic RNA and its role in viral RNA replication. RNA 2003; 9: 331-345. Links
- Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science 2001; 291: 1959-1962. Links
- Kieft JS, Zhou K, Grech A, Jubin R, Doudna JA. Crystal structure of an RNA tertiary domain essential to HCV IRES-mediated translation initiation. Nat Struct Biol 2002; 9: 370-374. Links
- Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, Chambers SP, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996; 87: 343-355. Links
- Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, et al. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 1996; 87: 331-342. Links
- Bianchi E, Pessi A. Inhibiting viral proteases: challenges and opportunities. Biopolymers 2002; 66: 101-114. Links
- Fischmann TO, Weber PC. Peptidic inhibitors of the hepatitis C virus serine protease within non-structural protein 3. Curr Pharm Des 2002; 8: 2533-2540. Links
- Borowski P, Schalinski S, Schmitz H. Nucleotide triphosphatase/helicase of hepatitis C virus as a target for antiviral therapy. Antiviral Res 2002; 55: 397-412. Links
- Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure Fold Des 1999; 7: 1353-1363. Links
- Caruthers JM, McKay DB. Helicase structure and mechanism. Curr Opin Struct Biol 2002; 12: 123-133. Links
- Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure Fold Des 1999; 7: 1417-1426. Links
- Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A 1999; 96: 13034-13039. Links
- Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol 1999; 6: 937-943. Links
- Bressanelli S, Tomei L, Rey FA, De Francesco R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J Virol 2002; 76: 3482-3492. Links
- Wang M, Ng KK, Cherney MM, Chan L, Yannopoulos CG, Bedard J, Morin N, et al. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J Biol Chem 2003; 278: 9489-9495. Links
- Flint M, Thomas JM, Maidens CM, Shotton C, Levy S, Barclay WS, McKeating JA. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J Virol 1999; 73: 6782-6790. Links
- Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, et al. Binding of hepatitis C virus to CD81. Science 1998; 282: 938-941. Links
- Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 2002; 21: 5017-5025. Links
- Pohlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, et al. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol 2003; 77: 4070-4080. Links
- Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A 1999; 96: 12766-12771. Links
- Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 2003; 197: 633-642. Links
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 2003; 100: 7271-7276. Links
- Thimme R, Bukh J, Spangenberg HC, Wieland S, Pemberton J, Steiger C, Govindarajan S, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A 2002; 99: 15661-15668. Links
- Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, Miller JL, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med 2000; 6: 578-582. Links
- Nascimbeni M, Mizukoshi E, Bosmann M, Major ME, Mihalik K, Rice CM, Feinstone SM, et al. Kinetics of CD4+ and CD8+ memory T-cell responses during hepatitis C virus rechallenge of previously recovered chimpanzees. J Virol 2003; 77: 4781-4793. Links
- Bassett SE, Guerra B, Brasky K, Miskovsky E, Houghton M, Klimpel GR, Lanford RE. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. HEPATOLOGY 2001; 33: 1479-1487. Links
- Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 2001; 15: 883-895. Links
- Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH, Liang TJ, et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol 2002; 169: 3447-3458. Links
- Kamal SM, Fehr J, Roesler B, Peters T, Rasenack JW. Peginterferon alone or with ribavirin enhances HCV-specific CD4 T-helper 1 responses in patients with chronic hepatitis C. Gastroenterology 2002; 123: 1070-1083. Links
- Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000; 275: 2247-2250. Links 61 Rockey DC. Hepatic fibrogenesis and hepatitis C. Semin Gastrointest Dis 2000; 11: 69-83. Links
- Friedman SL. Liver fibrosis - from bench to bedside. J Hepatol 2003; 38( suppl 1): S38-S53. Links
- Friedman SL. Hepatic stellate cells. Prog Liver Dis 1996; 14: 101-130. Links
- Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A 1985; 82: 8681-8685. Links
- Pestka S. The human interferon alpha species and receptors. Biopolymers 2000; 55: 254-287. Links
- Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol 1997; 15: 563-591. Links 67 Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 2003; 4: 63-68. Links
- Blatt LM, Davis JM, Klein SB, Taylor MW. The biologic activity and molecular characterization of a novel synthetic interferon-alpha species, consensus interferon. J Interferon Cytokine Res 1996; 16: 489-499. Links
- Tong MJ, Reddy KR, Lee WM, Pockros PJ, Hoefs JC, Keeffe EB, Hollinger FB, et al. Treatment of chronic hepatitis C with consensus interferon: a multicenter, randomized, controlled trial. HEPATOLOGY 1997; 26: 747-754. Links
- Chang CC, Chen TT, Cox BW, Dawes GN, Stemmer WP, Punnonen J, Patten PA. Evolution of a cytokine using DNA family shuffling. Nat Biotechnol 1999; 17: 793-797. Links
- Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr, Haussinger D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347: 975-982. Links
- Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358: 958-965. Links
- Pepinsky RB, LePage DJ, Gill A, Chakraborty A, Vaidyanathan S, Green M, Baker DP, et al. Improved pharmacokinetic properties of a polyethylene glycol-modified form of interferon-beta-1a with preserved in vitro bioactivity. J Pharmacol Exp Ther 2001; 297: 1059-1066. Links \74 Osborn BL, Olsen HS, Nardelli B, Murray JH, Zhou JX, Garcia A, Moody G, et al. Pharmacokinetic and pharmacodynamic studies of a human serum albumin-interferon-alpha fusion protein in cynomolgus monkeys. J Pharmacol Exp Ther 2002; 303: 540-548. Links
- Dockrell DH, Kinghorn GR. Imiquimod and resiquimod as novel immunomodulators. J Antimicrob Chemother 2001; 48: 751-755. Links
- Goldstein D, Hertzog P, Tomkinson E, Couldwell D, McCarville S, Parrish S, Cunningham P, et al. Administration of imiquimod, an interferon inducer, in asymptomatic human immunodeficiency virus-infected persons to determine safety and biologic response modification. J Infect Dis 1998; 178: 858-861. Links
- Strominger NL, Brady R, Gullikson G, Carpenter DO. Imiquimod-elicited emesis is mediated by the area postrema, but not by direct neuronal activation. Brain Res Bull 2001; 55: 445-451. Links
- Lau JY, Tam RC, Liang TJ, Hong Z. Mechanism of action of ribavirin in the combination treatment of chronic HCV infection. HEPATOLOGY 2002; 35: 1002-1009. Links
- Watson J. Prospects for hepatitis C virus therapeutics: levovirin and viramidine as improved derivatives of ribavirin. Curr Opin Investig Drugs 2002; 3: 680-683. Links
- Wu JZ, Walker H, Lau JY, Hong Z. Activation and deactivation of a broad-spectrum antiviral drug by a single enzyme: adenosine deaminase catalyzes two consecutive deamination reactions. Antimicrob Agents Chemother 2003; 47: 426-431. Links
- Sintchak MD, Nimmesgern E. The structure of inosine 5-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology 2000; 47: 163-184. Links
- Maxamine. Histamine dihydrochloride. Drugs R D 1999; 2: 274-275. Links 83 Billich A. Thymosin alpha1. SciClone Pharmaceuticals. Curr Opin Investig Drugs 2002; 3: 698-707. Links
- McHutchison JG, Patel K. Future therapy of hepatitis C. HEPATOLOGY 2002; 36: S245-S252. Links
- Pockros PJ, Patel K, O'Brien C, Tong M, Smith C, Rustgi V, Carithers RL, et al. A randomized, double-blind, placebo-controlled, multicenter study of recombinant human interleukin-12 for the treatment of chronic hepatitis C virus infection in patients non-responsive to previous treatment with interferon-alfa with or without ribavirin. HEPATOLOGY 2003; 37: 1368-1374. Links
- Hanecak R, Brown-Driver V, Fox MC, Azad RF, Furusako S, Nozaki C, Ford C, et al. Antisense oligonucleotide inhibition of hepatitis C virus gene expression in transformed hepatocytes. J Virol 1996; 70: 5203-5212. Links 87 Zhang H, Hanecak R, Brown-Driver V, Azad R, Conklin B, Fox MC, Anderson KP. Antisense oligonucleotide inhibition of hepatitis C virus (HCV) gene expression in livers of mice infected with an HCV-vaccinia virus recombinant. Antimicrob Agents Chemother 1999; 43: 347-353. Links
- McHutchison JG, Pockros PJ, Patel K, Nyberg L, Yu RZ, Kwoh TJ, Dorr FA. A phase 1b dose escalation trial of ISIS 14803, an antisense inhibitor of HCV, in patients with chronic HCV: final report. HEPATOLOGY 2002; 36: 303A. Links 89 Gordon SC, Bacon BR, Jacobson IM, Shiffman MI, Afdhal NH, McHutchison JG, Kwoh TJ, et al. A phase II, 12-week study of ISIS 14803, an antisense inhibitor of HCV for the treatment of chronic hepatitis C. HEPATOLOGY 2002; 36: 362A. Links
- Macejak DG, Jensen KL, Pavco PA, Phipps KM, Heinz BA, Colacino JM, Blatt LM. Enhanced antiviral effect in cell culture of type 1 interferon and ribozymes targeting HCV RNA. J Viral Hepat 2001; 8: 400-405. Links
- Hannon GJ. RNA interference. Nature 2002; 418: 244-251. Links
- Durantel D, Branza-Nichita N, Carrouee-Durantel S, Butters TD, Dwek RA, Zitzmann N. Study of the mechanism of antiviral action of iminosugar derivatives against bovine viral diarrhea virus. J Virol 2001; 75: 8987-8998. Links
- Pavlovic D, Neville DCA, Argaud O, Blumberg B, Dwek RA, Fischer WB, Zitzmann N. The hepatitis C virus p7 protein forms an ion channel that is inhibited by long-alkyl-chain iminosugar derivatives. Proc Natl Acad Sci U S A 2003; 100: 6104-6108. Links
- De Francesco R, Rice CM. New therapies on the horizon for hepatitis C: are we close? Clin Liver Dis 2003; 7: 211-242. Links
- Lamarre D, Bailey M, Bolger G, Cameron D, Cartier M, Faucher AM, Goudreau N, et al. The discovery of BILN 2061. An orally bioavailable small molecule inhibitor of the HCV serine protease and a promising antiviral for the treatment of hepatitis C. HEPATOLOGY 2002; 36: 279A. Links
- Benhamou Y, Hinrichsen H, Sentjens R, Reiser M, Manns MP, Forns X, Avendano C, et al. Safety, tolerability and antiviral effect of BILN 2061, a novel HCV serine protease inhibitor, after oral treatment over 2 days in patients with chronic hepatitis C, genotype 1, with advanced liver fibrosis. HEPATOLOGY 2002; 36: 304A. Links
- Hinrichsen H, Benhamou Y, Reiser M, Sentjens R, Wedemeyer H, Calleja JL, Forns X, et al. First report on the antiviral efficacy of BILN 2061, a novel oral serine protease inhibitor, in patients with chronic hepatitis C genotype 1. HEPATOLOGY 2002; 36: 379A. Links
- Walker MP, Hong Z. HCV RNA-dependent RNA polymerase as a target for antiviral development. Curr Opin Pharmacol 2002; 2: 534-540. Links
- Farci P, Alter HJ, Wong DC, Miller RH, Govindarajan S, Engle R, Shapiro M, et al. Prevention of hepatitis C virus infection in chimpanzees after antibody-mediated in vitro neutralization. Proc Natl Acad Sci U S A 1994; 91: 7792-7796. Links
- Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A 1996; 93: 15394-15399. Links
- Krawczynski K, Fattom A, Culver D, Kamili S, Spelbring J, Basham L, Sapan C, et al. Passive transfer of anti-HCV in chronic and acute HCV infection in chimpanzees: trials of experimental immune treatment. HEPATOLOGY 1999; 30: 423A. Links
- Krawczynski K, Fattom A, Spelbring J, Naso R, Sapan C, Purdy M, Basham L, et al. Early termination of HCV infection by passive anti-HCV transfer in experimentally infected chimpanzees. HEPATOLOGY 1998; 28: 398A. Links
- Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol 2001; 75: 5550-5558. Links
- Depraetere S, Van Kerschaever E, Van Vlierberghe H, Elewaut A, Brouwer JT, Niesters HG, Schalm SW, et al. Long term response to interferon treatment in chronic hepatitis C patients is associated with a significant reduction in anti-E1 envelope antibody titers. J Med Virol 2000; 60: 126-132. Links
- Baumert TF, Wellnitz S, Aono S, Satoi J, Herion D, Tilman Gerlach J, Pape GR, et al. Antibodies against hepatitis C virus-like particles and viral clearance in acute and chronic hepatitis C. HEPATOLOGY 2000; 32: 610-617. Links
- Leroux-Roels G, Depla E, Hulstaert F, De Smedt J, Desombere I, Maertens G. A candidate therapeutic vaccine for chronic hepatitis C infection based on envelope 1 protein: tolerability and immunogenicity in healthy adult volunteers. HEPATOLOGY 2000; 34: 449A
|
|
| |
| |
|
|
|