| |
Fosamprenavir (908, LEXIVA) Antivirial Activity Report
|
| |
| |
Reported by Jules Levin
This report contains results from two studies comparing fosamprenavir once and twice daily to nelfinavir (Viracept) in treatment-naïve patients; and results from a study comparing fosamprenavir twice daily to Kaletra in treatment-experienced patients. Resistance and cross-resistance is discussed below, as well as Warnings and precautions regarding fosamprenavir. Information in this report excerpted from FDA Lexiva Package Insert.
Effects of Food on Oral Absorption: LEXIVA Tablets may be taken with or withoutFood. Administration of a single 1,400-mg dose of LEXIVA in the fed state (standardized high-fat meal: 967 kcal, 67 grams fat, 33 grams protein,58 grams carbohydrate) compared to the fasted state was associated with no significant changes in amprenavir Cmax, Tmax, or AUC.
RESISTANCE: HIV-1 isolates with a decreased susceptibility to amprenavir have been selected in vitro and obtained from patients treated with fosamprenavir. Genotypic analysis of isolates from amprenavir-treated patients showed mutations in the HIV-1 protease gene resulting in amino acid substitutions primarily at positions V32I, M46I/L, I47V, I50V, I54L/M, and I84V, as well as mutations in the p7/p1 and p1/p6 Gag and Gag-Pol polyprotein precursor cleavage sites. Some of these amprenavir resistance-associated mutations have also been detected in HIV-1 isolates from antiretroviral-naive patients treated with LEXIVA.
Of the 488 antiretroviral-naive patients treated with LEXIVA or LEXIVA/ritonavir, 61 patients (29 receiving LEXIVA and 32 receiving LEXIVA/ritonavir) with virological failure (plasma HIV-1 RNA>1,000 copies/mL on 2 occasions on or after Week 12) were genotyped. Five of the 29 antiretroviral-naive patients (17%) receiving LEXIVA without ritonavir had evidence of genotypic resistance to amprenavir: I54L/M (n = 2), I54L + L33F (n = 1), V32I + I47V (n = 1), and M46I + I47V (n = 1).No amprenavir-associated mutations were detected in antiretroviral-naive patients treated with LEXIVA/ritonavir. Similarly to Kaletra patients experiencing viral failure to a Lexiva/r regimen did not show 908 genotypic mutations.
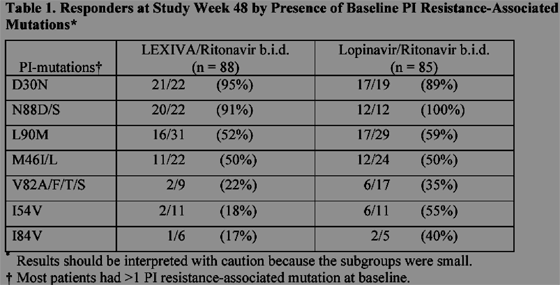
Cross-Resistance: Varying degrees of cross-resistance among HIV-1 protease inhibitors have been observed. An association between virologic response at 48 weeks (HIV-1 RNA level <400 copies/mL) and PI-resistance mutations detected in baseline HIV-1 isolates from PI-experienced patients receiving LEXIVA/ritonavir twice daily (n = 88), or lopinavir/ritonavir twice daily (n = 85) in study APV30003 is shown in Table 1. The majority of subjects had previously received either one (47%) or 2 PIs (36%), most commonly nelfinavir (57%) and indinavir (53%). Out of 102 subjects with baseline phenotypes receiving twice-daily LEXIVA/ritonavir, 54% (55) had resistance to at least one PI with 98% (54) of those having resistance to nelfinavir. Out of 97 subjects with baseline phenotypes in the lopinavir/ritonavirarm, 60% (58) had resistance to at least one PI with 97% (56) of those having resistance to nelfinavir.
In the table below, when the I54V, I84V, or V82A/F/T/S mutations were present response rates were better to Kaletra. When nelfinavir associated D30N mutation was present response rates were similar, 95% for Lexiva/r vs 89% for Kaletra, suggesting that nelfinavir viral failure may be relatively easier to respond. When the L90M mutation was present 52% responded to Lexiva/r vs 59% to Kaletra.
|
|
| |
| |
 |
|
| |
| |
The virologic response based upon baseline phenotype was assessed. Baseline isolates from PI-experienced patients responding to LEXIVA/ritonavir twice daily had a median shift in susceptibility to amprenavir relative to a standard wild-type reference strain of 0.7 (range: 0.1 to 5.4, n = 62), and baseline isolates from individuals failing therapy had a median shift in susceptibility of 1.9 (range: 0.2 to 14, n = 29). Because this was a select patient population, these data do not constitute definitive clinical susceptibility break points. Additional data areneeded to determine clinically relevant break points for LEXIVA.
Isolates from 15 of the 20 patients receiving twice-daily LEXIVA/ritonavir and experiencing virologic failure/ongoing replication were subjected to genotypic analysis. The following amprenavir resistance-associated mutations were found either alone or in combination: V32I, M46I/L, I47V, I50V, I54L/M, and I84V.
LEXIVA is indicated in combination with other antiretroviral agents for the treatment of HIV infection in adults.
The following points should be considered when initiating therapy withLEXIVA/ritonavir in protease inhibitor-experienced patients (see Description of Clinical Studies).
- The protease inhibitor-experienced patient study was not large enough to reach a definitive conclusion that LEXIVA/ritonavir and lopinavir/ritonavir are clinically equivalent.
- Once-daily administration of LEXIVA plus ritonavir is not recommended for protease inhibitor-experienced patients.
Description of Clinical Studies: Therapy-Naive Patients: Study APV30001:
Lexiva 1400 mg twice daily vs Nelfinavir 1250 mg twice daily
APV30001 was a randomized, open-label study, comparing treatment with LEXIVA Tablets (1,400 mg twice daily) versus nelfinavir (1,250 mg twice daily) in 249 antiretroviral treatment-naive patients. Both groups of patients also received abacavir (300 mg twice daily) and lamivudine (150 mg twice daily).
The mean age of the patients in this study was 37 years (range 17 to 70 years), 69% of the patients were males, 20% were CDC Class C (AIDS), 24% were Caucasian, 32% were black, and 44% were Hispanic. At baseline, the median CD4+ cell count was 212 cells/mm3 (range: 2 to 1,136 cells/mm3; 18% of patients had a CD4+ cell count of <50 cells/mm3 and 30% were in the range of 50 to <200 cells/mm3). Baseline median HIV-1 RNA was 4.83 log10 copies/mL (range:1.69 to 7.41 log10 copies/mL; 45% of patients had >100,000 copies/mL).
166 patients received Lexiva and 83 nelfinavir.
--RESPONDERS:
<400 copies/ml--66% Lexiva -- 52% NFV
<50 copies/ml—57% Lexiva -- 42% NFV
--VIRAL FAILURE- 19% Lexiva, 32% NFV
rebound - 16% Lexiva, 19% NFV
never suppressed through week 48- 3% Lexiva, 13% NFV
--Discontinued due to adverse events- 4% Lexiva, 2% NFV
Proportions of Responders by Baseline Viral Load
<100,000 copies/ml:
--<400 copies/ml:
65% on Lexiva 1400mg bid
65% on nelfinavir 1250mg bid
>100,000 copies/ml:
--<400 copies/ml
67% on Lexiva 1400 mg bid
36% on nelfinavir 1250 mg bid
Fosamprenavir (908) 1400 mg Once Daily + ritonavir 200mg once daily vs nelfinavir 1250 mg bid
Study APV30002: APV30002 was a randomized, open-label study, comparing treatment with LEXIVA Tablets (1,400 mg once daily) plus ritonavir (200 mg once daily) versus nelfinavir (1,250 mg twice daily) in 649 treatment-naive patients. Both treatment groups also received abacavir (300 mg twice daily) and lamivudine (150 mg twice daily). The mean age of the patients in this study was 37 years (range 18 to 69 years), 73% of the patients were males, 22% were CDC Class C, 53% were Caucasian, 36% were black, and 8% were Hispanic. At baseline, the median CD4+ cell count was 170 cells/mm3 (range: 1 to 1,055 cells/mm3; 20% of patients had a CD4+ cell count of <50 cells/mm3 and 35% were in therange of 50 to <200 cells/mm3). Baseline median HIV-1 RNA was 4.81 log10 copies/mL (range: 2.65 to 7.29 log10 copies/mL; 43% of patients had >100,000 copies/mL).
322 patients taking 908 1400 mg once daily + ritonavir 200mg once daily, and 327 patients received nelfinavir 1250 mg bid.
RESPONDERS
<400 copies/ml
--69% receiving fosamprenavir
--68% receiving nelfinavir
<50 copies
--58% receiving fosamprenavir
--55% receiving nelfinavir
virologic failures: 6% 908, 16% nelfinavir
--rebound: 5% 908, 8% NFV
--never suppressed through 48 weeks: 1% 908, 8% NFV
disct due to adverse events: 9% 908, 6% NFV
Proportions of Responders Through week 48 by Baseline Viral Load
<100,000 copies/ml
--<400 c/ml
72% taking Lexiva/r
73% NFV
>100,000
66% Lexiva/r
64% NFV
Kaletra vs 908 + ritonavir twice daily and once daily
Protease Inhibitor-Experienced Patients: Study APV30003: APV30003 was arandomized, open-label, multicenter study comparing 2 different regimens of LEXIVA plus ritonavir (LEXIVA Tablets 700 mg twice daily plus ritonavir 100 mg twice daily or LEXIVA Tablets 1,400 mg once daily plus ritonavir 200 mg once daily) versus lopinavir/ritonavir (400 mg/100 mg twice daily) in 315 patients who had experienced virologic failure to 1 or 2 prior protease inhibitor-containing regimens.
The mean age of the patients in this study was 42 years (range 24 to 72 years), 85% were male, 33% were CDC Class C, 67% were Caucasian, 24% were black, and 9% were Hispanic. The median CD4+ cell count at baseline was 263 cells/mm3 (range: 2 to 1,171 cells/mm3). Baseline median plasma HIV-1 RNA level was 4.14 log10 copies/mL (range: 1.69 to 6.41 log10 copies/mL).
The median durations of prior exposure to NRTIs were 257 weeks for patients receiving LEXIVA/ritonavir twice daily (79% had >=3 prior NRTIs) and 210 weeks for patients receiving lopinavir/ritonavir (64% had >=3 prior NRTIs). The median durations of prior exposure to protease inhibitors were 149 weeks for patients receiving LEXIVA/ritonavir twice daily (49% received >=2 prior PIs) and 130 weeks for patients receiving lopinavir/ritonavir (40% received >=2 prior PIs).
The time-averaged changes in plasma HIV-1 RNA from baseline (AAUCMB) at 48 weeks (the endpoint on which the study was powered) were ñ1.4 log10 copies/mL for twice-daily LEXIVA/ritonavir and ñ1.67 log10 copies/mL for the lopinavir/ritonavir group.
The proportions of patients who achieved and maintained confirmed HIV-1 RNA<400 copies/mL (secondary efficacy endpoint) were 58% with twice-daily LEXIVA/ritonavir and 61% with lopinavir/ritonavir (95% CI for the difference -16.6, 10.1). The proportions of patients with HIV-1 RNA <50 copies/mL with twice-daily LEXIVA/ritonavir and with lopinavir/ritonavir were 46% and 50%, respectively (95% CI for the difference -18.3, 8.9). The proportions of patients who were virologic failures were 29% with twice-daily LEXIVA/ritonavir and 27% with lopinavir/ritonavir.
The frequency of discontinuations due to adverse events and other reasons, and deaths were similar between treatment arms.
Through 48 weeks of therapy, the median increases from baseline in CD4+ cell counts were 81 cells/mm3 with twice-daily LEXIVA/ritonavir and 91 cells/mm3 with lopinavir/ritonavir.
This study was not large enough to reach a definitive conclusion that LEXIVA/ritonavir and lopinavir/ritonavir are clinically equivalent.
Once-daily administration of LEXIVA plus ritonavir is not recommended for protease inhibitor-experienced patients. Through Week 48, 50% and 37% of patients receiving LEXIVA/ritonavir once daily had plasma HIV-1 RNA <400 copies/mL and <50 copies/mL, respectively.
WARNINGS
Severe and life-threatening skin reactions, including Stevens-Johnson syndrome, have occurred in patients treated with amprenavir (see ADVERSE REACTIONS). Acute hemolytic anemia has been reported in a patient treated with amprenavir.
Serious and/or life-threatening drug interactions could occur between LEXIVA and amiodarone, lidocaine (systemic), tricyclic antidepressants, and quinidine. Concentration monitoring of these agents is recommended if these agents are used concomitantly with LEXIVA.
Severe and life-threatening skin reactions, including Stevens-Johnson syndrome, have occurred in patients treated with amprenavir (see ADVERSE REACTIONS). Acute hemolytic anemia has been reported in a patient treated with amprenavir.
Rifampin should not be used in combination with LEXIVA because it reduces plasma concentrations of amprenavir by about 90%. The effect of rifampin on amprenavir concentrations when rifampin is administered with LEXIVA plus ritonavir is not known.
Concomitant use of LEXIVA and St. John's wort (hypericum perforatum) or products containing St. John's wort is not recommended. Coadministration of protease inhibitors, including LEXIVA, with St. John's wort is expected to substantially decrease protease inhibitor concentrations and may result in suboptimal levels of amprenavir and lead to loss of virologic response and possible resistance to LEXIVA or to the class of protease inhibitors.
Concomitant use of LEXIVA with lovastatin or simvastatin is not recommended. Caution should be exercised if HIV protease inhibitors, including LEXIVA, are used concurrently with other HMG-CoA reductase inhibitors that are also metabolized by the CYP3A4 pathway (e.g., atorvastatin). The risk of myopathy, including rhabdomyolysis, may be increased when HIV protease inhibitors, including LEXIVA, are used in combination with these drugs.
Particular caution should be used when prescribing phosphodiesterase (PDE5) inhibitors for erectile dysfunction (e.g., sildenafil or vardenafil) in patients receiving protease inhibitors, including LEXIVA. Coadministration of a protease inhibitor with a PDE5 inhibitor is expected to substantially increase the PDE5 inhibitor concentration and may result in an increase in PDE5 inhibitor-associated adverse events, including hypotension, visual changes, and priapism: Drug Interactions and Information for Patients, and the complete specificPDE5 inhibitor prescribing information).
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, andhyperglycemia have been reported during post-marketing surveillance in HIV-infected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarilyduring clinical practice, estimates of frequency cannot be made and causal relationships between protease inhibitor therapy and these events have not been established.
PRECAUTIONS
Sulfa Allergy: LEXIVA should be used with caution in patients with a known sulfonamide allergy. Fosamprenavir contains a sulfonamide moiety. The potential for cross-sensitivity between drugs in the sulfonamide class and fosamprenavir is unknown. In a clinical study of LEXIVA used as the sole protease inhibitor, rash occurred in 2 of 10 patients (20%) with a history of sulfonamide allergy compared with 42 of 126 patients (33%) with no history of sulfonamide allergy. In 2 clinical studies of LEXIVA plus low-dose ritonavir, rash occurred in 8 of 50 patients (16%) with a history of sulfonamide allergy compared with 50 of 412 patients (12%) with no history of sulfonamide allergy.
|
|
| |
| |
|
|
|