| |
Recommendations for the Nonclinical Development of Topical Microbicides for Prevention of HIV Transmission: An Update
|
| |
| |
JAIDS Journal of Acquired Immune Deficiency Syndromes: Volume 36(1) 1 May 2004
Lard-Whiteford, Sheryl L. PhD*#; Matecka, Dorota†#; O'Rear, Julian J.†#; Yuen, Ita S.†#; Litterst, Charles‡#; Reichelderfer, Patricia§#; for the International Working Group on Microbicides
From *Center for Biologics Evaluation and Research, U.S. Food and Drug Administration, Rockville, MD, †Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Rockville, MD, ‡National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, and §National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD.
#The views expressed are those of the authors and do not necessarily represent those of the U.S. Food and Drug Administration and National Institute of Health.||The members of the International Working Group on Microbicides are listed under Acknowledgments.
The International Working Group on Microbicides endorsed this article, but it does not necessarily reflect the policies of individual members or their respective agencies.
Abstract
Abstract: The development of methods to prevent HIV infection is critical to curbing the rising epidemic. Topical microbicides represent a potential new strategy for reduction of HIV transmission. The purpose of this article is to update and expand upon the nonclinical recommendations of a previously published document on the development of microbicides prepared by the International Working Group on Microbicides. The nonclinical studies discussed here represent general concepts and regulatory considerations that are pertinent to the development of topical microbicides for prevention or reduction of HIV transmission. Essential early steps in product development include the determination of antiviral activity, cytotoxicity, mechanism of action, pathways to resistance, and cross-resistance to approved drugs. Other parameters to consider include activity against vaginal microflora and pathogens that cause sexually transmitted diseases. Before and during clinical trials, nonclinical data on toxicology and pharmacokinetics should be obtained. Finally, product quality issues, including microbicide formulation characteristics, interaction with other products, and stability, should be addressed.
The incidence of HIV infection is increasing among women worldwide. Although barrier methods such as the condom may block HIV transmission when used appropriately, there is a need for female-controlled methods to prevent infection. Topical microbicides (ie, vaginal or rectal products that interfere with HIV infection) may be a means by which women can protect themselves. As such, topical microbicide development constitutes one of the cornerstones of the prevention science agenda for HIV infection.
Both nonclinical and clinical developments of microbicides present unique challenges. In an effort to facilitate their development, the International Working Group on Microbicides (IWGM) published recommendations regarding the design of both nonclinical and clinical testing regimens for these products. The IWGM is an unofficial group consisting of representatives from diverse governmental and nongovernmental agencies and organizations who work in the area of microbicide development. The purpose of the group is to facilitate the development, production, and distribution of safe, effective, and affordable microbicides to prevent or reduce the sexual transmission of HIV and other sexually transmitted infections (STIs). The recommendations, originally published in 1996, addressed both clinical and nonclinical development processes. With the rapid advances made in the laboratory sciences and the increased complexity of clinical trial design issues, the group decided to issue separate documents. An updated version of the recommendations pertaining to the clinical development phase was published in 2001. The present article summarizes the recommendations of the group regarding the nonclinical development process for microbicides to prevent vaginal transmission of HIV.
The nonclinical development of a potential microbicide encompasses studies in several areas, which include microbiology, pharmacology/toxicology, and chemistry, manufacturing, and controls (CMC). The main purpose of these studies is to ensure that the estimated risk/benefit profile of a novel pharmaceutical is reasonable for the proposed indication before it is introduced into humans in clinical trials. Data from in vitro and animal model studies should provide some evidence of biological plausibility (ie, an indication that the candidate product has a type and level of activity that could be expected to provide some protection against HIV infection in vivo under the proposed conditions for use). These studies can also indicate whether a microbicide has any effects on the endogenous microflora in the female genital tract, a property that could influence its activity profile in vivo. The results of both in vitro and animal pharmacokinetic and toxicological studies provide data on the margin of safety of the drug as well as potential toxicological signals that need to be monitored in the clinical trials. The goal of CMC studies for any pharmaceutical is to ensure the identity, purity, quality, and strength of both the drug substance (active pharmaceutical ingredient) and the drug product. There are also issues specific to the development of the topical microbicide formulation that may require special attention (eg, additional testing and controls).
During clinical development, nonclinical studies can serve as a signal generator and can complement areas of safety and activity that cannot be studied in humans. For example, it is unethical to study the effects of a drug on the developing human fetus and impractical to study the carcinogenic potential of a drug in humans, both of which can be studied in animals.
Designs of many recommended studies listed below have been described in documents published by the International Conference on Harmonization (ICH) with the United States, Europe, and Japan being the major participants. Therefore, they represent internationally recognized guidance for product development. Whenever possible, examples of potentially useful assay systems and animal models published in recently peer-reviewed journals are referenced. These references are intended to provide the reader with additional sources of information on product development options and relevant assay systems. The proceedings of the first international meeting on microbicides, Microbicides 2000, may also be helpful. Additional examples of relevant assay methods and study designs may be available from the literature pertaining to nonclinical evaluation of new HIV therapeutics and vaccines, where peer-reviewed publications are more prevalent. Discussions with the appropriate regulatory agency early in the development process are highly recommended.
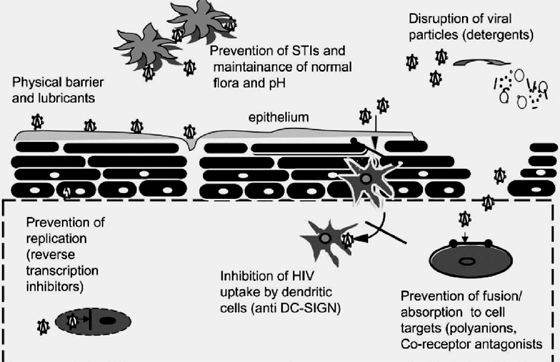
The recommendations discussed in this article represent a generalized scheme for the nonclinical development of topical microbicides to prevent or reduce vaginal transmission of HIV. The final development plan for a given product will follow this general outline but may be adjusted to address the specific characteristics of the product under development and its proposed indication for use. For example, there are many potential mechanistic approaches currently under consideration in microbicide development, as illustrated in Figure 1.
They include maintenance of normal vaginal pH and/or flora that are inhibitory to infection, establishment of a physical barrier to prevent infection, disruption of viral particles, and inhibition of attachment, fusion of the target cell and viral membranes, or reverse transcription. Although all products will be expected to have data demonstrating antiviral activity, the specific types of nonclinical activity studies that are performed to fully evaluate a candidate product should reflect the proposed mechanism of action of that specific product. Some promising microbicide candidates may also have activity against a variety of other STIs in addition to HIV infection. Products under development for a more general STI prevention indication require specific activity studies directed at the other pathogens as well, expanding the scope of the nonclinical activity assessment. Finally, some products may be considered for both rectal and vaginal use. Additional nonclinical studies that are not specifically addressed here will be required to address the unique scientific and regulatory challenges associated with the development of rectal use products.
|
|
| |
| |
 |
|
| |
| |
FIGURE 1. Potential sites at which a microbicide might act to prevent or reduce penetration and/or infection. A topical microbicide candidate may operate by establishing a physical barrier to block HIV and infected cells from entering into the body by maintaining normal vaginal pH and/or microflora that are inhibitory to infection or by acting against other STI pathogens that are cofactors for HIV infection. It can also be a detergent or have a detergent-like property to disrupt viral particles or contain an active ingredient specifically designed to inhibit viral uptake by dendritic cells (anti DC-SIGN [dendritic cell-specific intercellular adhesion molecule grabbing nonintegrin]), fusion of the target cell and viral membranes (eg, polyanions and coreceptor antagonists), or reverse transcription to prevent or reduce HIV transmission. A candidate microbicide may also act by more than one of the proposed mechanisms of action. (Adapted with permission from Dr. Robin Shattock.)
EVALUATION OF ANTIMICROBIAL ACTIVITY
Cell-Based Assays
The nonclinical evaluation of antiviral activity using cell-based assays is an essential part of the development process for a potential microbicide. In vitro cell-based assays are routinely conducted with the active pharmaceutical ingredient(s), either alone or in combination with other potentially active agents. The primary goal of this assessment is to define the full range of antiviral activity of the candidate drug substance and to obtain preliminary information on antiviral activity under different conditions. These data, which are typically derived from a combination of in vitro model systems, should support the rationale for clinical testing in humans by providing clear evidence of a potentially beneficial effect at levels that can be achieved in vivo with minimal or, preferably, no toxicity. Nonclinical antiviral activity and cytotoxicity data can also be used to guide the selection of an appropriate dose (or dose range) for early clinical trials and to support the process of formulation development.
Types of Assays
The evaluation of microbicide antiviral activity is often a stepwise process that proceeds from basic in vitro screening tests to a more detailed analysis of product activity including dose/response relationships, spectrum of activity, and mechanism of action. Screening programs typically employ a highly reproducible cell-based assay with an automated readout of viral replication to rapidly evaluate a large number of compounds for evidence of antiviral activity. In many cases, only a single virus or cell type at one infectious dose of a standard laboratory strain and 1 or 2 dilutions of the compound are tested. Even with this limited focus, the primary screening should include both cell-associated and cell-free virus. Although there is no consensus on the types of viruses that should be used for large-scale screening purposes, tests of viruses that use the CXCR4 coreceptor and viruses that use the CCR5 co-receptor for entry are generally preferred. Viruses that are highly cytopathic cannot be readily incorporated into some existing assays; therefore, care must be taken when selecting the virus/cell combination for a screening test. It is important to note that most large-scale screening programs were established to identify potential therapeutic compounds directed against HIV. Although these programs can provide a useful source of candidate compounds for microbicide development, they tend to focus on agents that block intracellular replication steps and should be viewed as one component of a more comprehensive screening and evaluation process.
Although active compounds may be identified through the use of various screening tests, candidates for further development require a more thorough in vitro evaluation to characterize the compounds' activity profile and define their mechanism of action. An essential step in this process is the establishment of the dose/response relationship using a broad range of relevant cell types and virus isolates. In general, in vitro cell-based assays fall into four formats: (1) virus inactivation assays for both cell-free and cell-associated virus; (2) epithelium-based assays (ME-180 cervical epithelial cells and/or GHOST X4/R5 human osteosarcoma cells); (3) peripheral blood mononuclear cell (PBMC) assays; and (4) binding and fusion assays. The types of assays used and the virus/cell combinations that are tested may vary somewhat with the compound under study. However, for all compounds under consideration for clinical testing, dose/response data should be obtained using primary human target cells and a wide range of clinical virus isolates as well as standard laboratory strains to provide adequate reference. Variables that should be addressed in these assays include the effect of exposure to the product before and after infection, the impact of an increasing multiplicity of infection, activity against the different HIV type 1 (HIV-1) clades, activity against clinical isolates derived from the lower reproductive tract, and the activity observed after pretreatment of virus versus pretreatment of cells. Excipients used in the drug product should be tested for potential enhancement of infectivity.
Primary human genital cell assays, studies conducted with continuous cell lines such as ME-180 cervical epithelial cells (a CD4- transformed cell line), and the newer ex vivo explant models may provide useful supporting data but cannot be used in place of dose/response curves in primary target cells. Data obtained using the ME-180 model is viewed as supplemental only, because the role of CD4- vaginal epithelial cells in the transmission of HIV in humans has not been established. Consequently, the practical value of activity data generated in this system is unknown. The model is also limited in that infection can only be accomplished with cell-associated virus. The explant model may provide a system that more closely reflects the vaginal environment. However, this type of culture system is relatively new; therefore, its potential utility and reproducibility in different hands and under different study conditions have not been fully explored.
Defining a Therapeutic Index
The nonclinical activity profile of the candidate compound should demonstrate that it is capable of interfering with the process of HIV infection over a range of concentrations that are not overtly cytotoxic. Antiviral activity usually results from disruption of the virus, blocking of viral attachment and/or membrane fusion, or inhibition of some other step in the virus life cycle such as reverse transcription or polyprotein processing. However, many of these agents could also affect normal cell viability, which may limit virus replication but can also lead to unwanted cellular and/or systemic toxicities. To characterize both aspects of this dose/response relationship, compounds should be tested simultaneously for antiviral activity and cytotoxicity over a wide range of drug and virus/cell concentrations. The goal is to identify the lowest effective antiviral concentration and the highest achievable level that also exhibits an acceptable cytotoxicity profile (ie, little or no cytotoxicity in cell culture). This relationship can be defined as the therapeutic index and is derived from the ratio of the agent concentration that reduces cell viability by 50% and the concentration that reduces infectivity by 50%. Presently, there is no consensus regarding the minimal level of antiviral activity that should be mediated by a potential HIV microbicide, but a clear distinction between antiviral and cytotoxic concentrations should be demonstrated. In some instances, the candidate microbicide may be cytotoxic (eg, low pH buffers), in which case the kinetics of inactivation of virus by the agent need to be determined to ascertain if the rate is consistent with the exposure times in actual use.
Selection of Virus and Cell Types
To optimize the value of an in vitro system for drug evaluation, a sensible approach would dictate that the system includes virus and cell types most likely to be involved in the infection event in vivo. Because the actual process by which HIV is transmitted through sexual contact has not been fully elucidated, it is necessary to test candidate compounds for antiviral activity using a variety of cell and virus combinations, focusing on cell types known to be targets for virus infection in vivo and the virus types believed to be the most likely to transmit infection. The potential for in vivo infection of T cells and macrophages has been well established; consequently, evaluation of the activity of candidate compounds using clinical virus isolates in both PBMC and macrophage cultures (not solely established cell lines) is considered to be an essential component of the nonclinical development process for potential microbicides. However, recent data suggest that dendritic cells may also serve as primary cell targets for infection in the course of sexual transmission. Dendritic cells have been shown to capture and internalize HIV-1 through DC-SIGN (dendritic cell-specific intercellular adhesion molecule grabbing nonintegrin, CD209) and possibly other cell surface receptors and then transfer the infectious virus to susceptible CD4+ T cells. As a result, an evaluation of candidate microbicides for activity in a primary dendritic cell model should also be considered, particularly for compounds that have the potential to influence virus binding and/or entry processes. Finally, because the viruses that are transmitted will be associated with semen or vaginal secretions, testing of compounds in the presence of these body fluids is critical.
Resistance
The emergence of resistance is a major impediment to the control of HIV transmission and long-term therapy. The major pathways to resistance for new agents should be identified by selection of resistant virus in vitro, which should be characterized genotypically and phenotypically. When resistance mutations arise in target proteins (reverse transcritpase and protease) or protein complexes (gp120 and gp41) for which drugs are already approved, cross-resistance should be assessed. Cross-resistance is not necessarily reciprocal. For example, if virus X is resistant to drug A and shows cross-resistance to drug B, virus Y, which is resistant to drug B, may still be sensitive to drug A. The activity of approved drugs against virus resistant to the candidate agent and the activity of the candidate agent against virus resistant to approved drugs should be evaluated.
Host Proteins
Recently, there has been much interest in the development of agents targeting host cell proteins (eg, CCR5 coreceptor). The antiviral activity of such agents should be established with all the common polymorphisms to ensure their efficacy. In addition, the agonistic/antagonistic effects on host cell function should be determined.
Animal Models and Surrogate Viruses
Although studies of product activity in nonhuman primate systems may also be performed, to date these studies have had limited utility in helping to decide which compounds should go forward into clinical trials. The relevance of these models to the process of sexual HIV transmission in humans is unknown, and the cost of doing studies that are large enough to provide statistically meaningful activity data may be prohibitive. However, the pigtailed macaque model may provide a useful tool for vaginal safety studies (eg, assessment of irritation potential or effects on normal flora). The female genital tract in this species has many characteristics in common with that in humans. Recently, murine models of HIV vaginal infection have been established. These models may prove useful in the future to test the activity of various microbicide formulations, but their utility for predicting clinically relevant antiviral activity has yet to be established.
Genital Tract Interaction
The normal vaginal environment has characteristics that help to keep it healthy, including a low pH that limits the replication and spread of potential pathogens. Resident microorganisms, including H2O2-producing lactobacilli, are largely responsible for maintaining the acidic pH of the vagina, but these beneficial bacteria may be sensitive to the antimicrobial effects of the candidate microbicide. The presence of abnormal flora (eg, bacterial vaginosis) and reduced numbers of H2O2-producing lactobacilli have been associated with an increased risk of HIV transmission. Given the importance of maintaining normal flora and pH, it is essential to determine the impact of the microbicide on H2O2-producing lactobacilli as part of the nonclinical development process. 37 In addition to available in vitro assays, the pigtailed macaque, which has genital and rectal flora similar to that in humans, has been used to assess changes in normal rectal and vaginal flora following administration of various products.
Effect on Other STIs
Many of the microbicide drug substances under development could influence the replication of other STI pathogens, in addition to HIV. Although most nonclinical activity studies conducted on compounds directed against HIV will be focused on that virus, it is important to determine what effect a candidate microbicide has upon other STIs (eg, inhibition or potentiation). Initial evaluations can be as simple as assessing the inhibitory concentration or growth potentiation effects in vitro against a panel of known pathogens such as Chlamydia trachomatis, Neisseria gonorrhoeae, herpes simplex virus type 2 (HSV-2), human papillomavirus (HPV), and organisms associated with bacterial vaginosis. More detailed studies that employ a variety of in vitro methods (eg, kinetics of inactivation at concentrations similar to the anticipated use) and animal models to assess activity should be considered when the drug product under development has broader-spectrum activity that may have clinical relevance. Murine models have been used to evaluate many potential microbicidal compounds for inhibition of STIs, and a pigtailed macaque model has been developed to evaluate activity against C. trachomatis. Animal models can be used to compare the relative activity of various compounds against various STIs and to determine the duration of that activity. The models have also been useful in the evaluation of safety as it relates to susceptibility (eg, enhancement of HSV-2 infection).
NONCLINICAL TOXICOLOGY AND PHARMACOKINETICS
Before introduction of a new chemical entity to humans, safety information is derived mainly from results of nonclinical toxicology and pharmacokinetic studies. Nonclinical pharmacokinetic studies provide information on the absorption (and also the extent of systemic exposure), distribution, metabolism, and excretion profile of a drug. Several aspects of toxicology also need to be investigated to estimate the margin of safety, establish potential toxicological signal(s), determine whether the toxicological signal(s) can be monitored in the clinical trials, and provide information for risk assessment. They include general toxicology studies, safety pharmacology studies, genetic toxicology studies, reproductive toxicology studies, and carcinogenicity studies. Potential irritation to the vagina and development of hypersensitivity or other types of immunotoxicity are particular to the topical microbicides and also should be monitored. Each of these areas will be discussed in this section.
It should also be mentioned that some of the topical microbicide candidates might come from already marketed products designed for intravaginal administration. For these types of products, no further nonclinical animal testing for toxicity will be needed. However, when a marketed product is reformulated for vaginal use, some bridging studies will be needed to address the toxicities that may be relevant to the new route of administration. Although not strictly required, it is desirable that the materials used in the animal toxicology studies are manufactured in compliance with the Current Good Manufacturing Practice (CGMP) requirements. It is also noteworthy that all pivotal nonclinical toxicology studies should use the product formulation that will be tested in clinical trials whenever possible, and all nonclinical toxicology studies should be carried out according to Good Laboratory Practice requirements.
Pharmacokinetic Studies
Quantification of the extent of drug substance absorption after administration of the formulated product into the vagina and/or rectum is essential in determining the strategy of nonclinical pharmacology/toxicology development. Pharmacokinetic studies in animals should monitor levels of drug substance in blood organs, and tissues that are relevant and critical and mass balance of administered-plus-recovered drug substance. Before conducting the pharmacokinetic studies, appropriate analytical methods of detection and quantification of the drug substance should be established. The absorption study is greatly facilitated if radiolabeled drug can be used and should use a volume and vehicle that assure complete spread throughout the vagina. The distribution and excretion pattern and metabolic profile of the drug formulation should also be investigated. If metabolites are formed, it is important to develop assays for their detection. The extent of metabolite absorption and systemic toxicity via the vagina should also be determined. The study should use the same formulation that will be used in clinical trials, whenever possible. The results of these investigations will be pivotal in determining which toxicology studies should be performed.
General Toxicology Studies
Nonclinical general toxicology studies are designed to characterize the toxicological profile of a drug by monitoring its effects on general health and behavior, weight changes, food consumption, hematology, serum chemistry, urinalysis parameters, and tissues and organs macroscopically and microscopically. These studies are generally conducted in a rodent and a nonrodent species, with the duration of the study and the frequency of dosing equal to or exceeding those of the human clinical trials. The doses studied should include one that induces no adverse effects, a dose that induces frank toxicity (a maximum tolerated dose), and an intermediate dose. The route of administration should normally be the same as that used in the clinical trials (ie, vaginal). However, the formulation and route of administration may limit the amount of systemic drug exposure so that achieving the maximum tolerated dose is not technically feasible. In such cases, alternative routes (ie, oral or parenteral) using drug substance may be warranted especially when systemic absorption in humans cannot be ruled out. If the drug substance is considered non toxic, the high dose for the subsequent toxicology studies can be a maximum feasible dose. The responsible regulatory agencies should be consulted early in the drug development program regarding the appropriate study design to explore the possible systemic toxicity profile of the drug. Plasma drug levels are usually monitored in these studies to determine the extent of systemic absorption after repeated drug exposure. With these data, a margin of safety for the drug product can be extrapolated from animal to human. In addition, the potential for cumulative adverse effects that may occur after repeated administration of the drug product is also explored. Sometimes, a recovery group may be incorporated into the study to determine the reversibility of an observed toxicity. Evaluation of immature red blood cells from the bone marrow for the presence of micronuclei may be included to monitor for potential genetic toxicity in the short-term studies. Long-term repeat dose toxicology studies where drug products are administered via vagina may not be required if the systemic toxicity profile of the drug substance is well understood.
Normally, the sequence of required general toxicology studies includes acute (dosing for a single day) studies followed by short-term (14 days to 3 months), subchronic (6 to 9 months), and chronic (2 years as in carcinogenicity studies) evaluation. In the acute toxicology studies, the drug substance is administered to animals via the oral or parenteral route. They are designed to provide a general impression of the toxicity of the tested substance, to define the dose at which toxicity becomes life threatening, and to identify the corresponding dose at which no adverse effects are noticed, when the chemical is administered systemically to animals. Drug products are usually used in all other general toxicity studies.
Genetic Toxicology Studies
The mutagenic potential of a pharmaceutical should be evaluated early in the vaginal microbicide development process using the standard ICH-recommended battery of tests. They are as follows: (1) a test for gene mutation in bacteria; (2) an in vitro test with cytogenetic evaluation of chromosomal damage with mammalian cells or an in vitro mouse lymphoma tk assay; and (3) an in vivo test for chromosomal damage using rodent hematopoietic cells. The Ames Salmonella test and the in vitro mammalian gene mutation assays should be conducted before the first clinical trial. These in vitro tests are rapid and inexpensive, and the recommended study design and cell strains can be found in the ICH guidance. An in vivo micro-nucleus test may be conducted during phase 1 clinical trials as a discrete study or as a component of the short-term general toxicology study where drug exposure in bone marrow is substantial.
Vaginal Irritation
A screening study for vaginal irritation (usually in rabbits) can be used to determine the vaginal irritation potential of a drug substance before extensive and expensive formulation development and animal model efficacy studies. Effects on vaginal tissues should be evaluated at several dose levels. If irritation is evident at dose levels near those targeted for clinical studies, further development should be reconsidered. Of much more critical importance, however, is the determination of the potential vaginal irritation produced by the formulated drug products. This is necessary not only for new products but also for products that are newly formulated or for common formulations that have undergone major modifications. The clinical formulation should be studied to establish that it does not cause irritation at concentrations targeted for clinical studies. A standard 10-day intravaginal administration in rabbits is recommended. The positive and vehicle controls should be used concomitantly in the study. Vaginas should be evaluated both macroscopically and microscopically for signs of irritation, inflammation, and ulceration. The scoring system usually follows that described by Eckstein et al. Recently, the pigtailed macaque (Macaca nemestrina) has also been used to study the potential of a topical microbicide formulation to cause vaginal irritation and its effects on the vaginal microflora. The results from such studies can be used to supplement those from the rabbit vaginal irritation study.
Safety Pharmacology Studies
For a topical microbicide candidate that has been shown to be absorbed systemically, the effects of the drug on the functions of the vital organs and systems at or above therapeutic exposure need to be assessed before introduction to humans. A core battery of safety pharmacology studies is recommended by the ICH S7A guidance to evaluate the drug effects on central nervous system, cardiovascular system, and respiratory system functions. If there are effects to other organ systems that are indicated by the results from the general toxicology studies, other safety pharmacology studies designed to further investigate the mechanism of action of the toxic effects may be necessary.
Reproductive Toxicology Studies
Because microbicides will be used by women of child-bearing age, the effect of the product on reproductive health and developing embryo/fetuses needs to be determined. The contraceptive potential of a microbicide candidate (ie, toxicity to the human sperm) should be studied. This may be evaluated using any in vitro protocol that studies a range of concentrations, including and exceeding that exhibiting in vitro anti-HIV activity.
The designs for reproductive toxicology studies have been agreed upon internationally. However, the timing of performance of each study may vary according to the requirements of the regulatory agency in each country. A segment I reproductive toxicology study to evaluate the potential toxicity to fertility and early embryonic development should be conducted in one species, usually rats. Reproductive toxicology studies to assess embryo-fetus development (segment II) should be conducted in a rodent and a nonrodent species, usually rats and rabbits. If the clinical route of administration does not allow a sufficient amount of the drug to be delivered to induce maternal toxicity, intravaginal administration can be used in one of the segment II reproductive toxicology studies, while the other study should employ an alternative (oral or parenteral) route of administration. Because the microbicides are expected to be administered intravaginally, with close proximity to the developing embryo and possible migration to other reproductive organs/tissues, it is prudent that the segment I and segment II reproductive toxicology studies be conducted as early in the drug development as possible, preferably before human exposure. The study to evaluate the effects of the drug on peri- and postnatal development (segment III) is conducted in a rodent species, usually rats, during phase 3 clinical trials.
Hypersensitivity and Other Safety Studies
Cutaneous immunogenicity can be studied to detect the potential of a microbicide to produce an immunogenic response with repeated administration to a cutaneous surface. This test has the ability to detect the potential for producing vaginal irritation by mechanisms that may be different from those operating in the standard vaginal irritation model. The local lymph node assay in mice may be conducted in lieu of the more traditional guinea pig sensitization assay.
Carcinogenicity Studies
Since microbicides are expected to be used chronically, their carcinogenicity potential needs to be evaluated and completed before market approval. This is usually done by dosing rats and mice for 2 years to determine if there is an evidence of tumorigenic effects. The route of administration is usually similar to that used for humans (ie, intravaginal application). In lieu of the traditional 2-year carcinogenicity study in mice, a short-term (usually 6 month in duration) alternative carcinogenicity assay can be employed. The most appropriate alternative assay for topical microbicides is probably the Tg.AC transgenic mouse model because it is expected that these types of products will be formulated for topical application and should be nongenotoxic. The high dose to be evaluated is selected based on one of the following criteria: the toxicity end points (the maximum tolerated dose derived from 3-month general toxicity studies), pharmacokinetic end points (25-fold over human systemic exposure), saturation of absorption, maximum feasible dose, pharmacodynamic end points, and limit dose. The carcinogenicity studies are usually conducted during phase 3 clinical trials, and the results need to be assessed for marketing approval of a pharmaceutical.
Numerous other nonclinical toxicology studies may be required before marketing approval, but these tests may be conducted while the compound is in clinical trials. In addition, studies of the irritation to rectal and buccal mucosa and penile irritation studies should be considered. In addition, photosensitization testing may be required if a drug or its metabolites are photosensitive and absorbed systemically. If the microbicide is poorly absorbed and/or highly bioadhesive, penile absorption studies may be requested. Other studies may be necessary depending on the completeness of the toxicology database and the toxicity profile of the drug product.
PRODUCT QUALITY CONSIDERATIONS
Microbicide products for vaginal use should meet the standard chemistry, manufacturing, and controls expectations in terms of their proper identification, quality, purity, and strength to assure the safety of the human subjects participating in clinical trials. These expectations apply to all aspects of the drug substance and the drug product manufacture (ie, synthesis or isolation, characterization, and purification, etc, of the drug substance as well as the composition, manufacturing process, in-process controls, packaging, stability, and specification, etc, of the drug product to assure its quality and reproducibility). In addition, microbicide products, as other pharmaceuticals, must be manufactured in compliance with CGMP requirements. Although the collection of information begins early in the drug development, when the drug substance is defined and characterized, many aspects of a manufacturing process of an investigational drug may be incomplete. Changes and improvements in synthetic/manufacturing processes, formulations, analytical methods, and specifications may take place as the drug development progresses to advanced phases of clinical studies. Subsequently, at the phase 3 stage, these aspects of the drug substance and the drug product manufacture should be finalized and be identical to those intended for the marketed product. Product for phase 3 trials should be adequately characterized to assure its sameness to the proposed commercial product (refer to all pertinent ICH guidances on quality).
Formulation Considerations
A major consideration in the development of a successful microbicide drug product is the design of a dosage form that would deliver a drug substance in a safe, effective, and consistent manner. In addition, the optimal microbicide vaginal formulation should have some important functional attributes, which would assist with product utility and user compliance. These attributes include prolonged retention and suitable distribution (formulation deployment in the vaginal tract, lack of toxicity and irritation, adequate dosage and volume of the formulation for the proposed dosing frequency, and compatibility with barrier contraceptives. An optimal vaginal drug product should not disrupt or sensitize the vaginal epithelium and should not enhance or support the growth of the specific pathogens (ie, Candida albicans, Escherichia coli, Pseudomonas aeruginosa, or Staphylococcus aureus). Each of these attributes can be influenced by the physical properties of the vaginal drug product, such as solubility in various pH environments, viscosity, stability, and so on. The release rate may be directly related to the solubility, hygroscopicity or solid-state form of the drug substance. Deployment of the drug product in the vagina depends on the rheological characteristics (eg, viscosity, elasticity, yield stress, etc) and adhesion of the vehicle.
Properties and attributes affecting the performance of a microbicide drug product should be carefully investigated in the formulation studies, where the composition of the vehicle containing inactive ingredients (such as oil, water, surfactants, preservatives, emulsifying and viscosity modifying agents, etc) is established. Attributes such as distribution and optimal volume have recently been under intensive scientific investigation. However, only limited data are available. Recently, new imaging techniques have been developed and are being studied as potential tools to measure these attributes and to design the optimal vaginal formulation. Formulation studies are also important, because they help in the subsequent establishment of a drug product specification (tests, acceptance criteria, and analytical procedures).
Tests for Vaginal Drug Products
Dosage forms used for vaginal administration include gels, creams, ointments, foams, suppositories, inserts, filled capsules, films, sponges, etc. The specification for the vaginal drug product should include standard attributes such as appearance, identity, impurities profile, content uniformity, homogeneity assurance, and assay for the content of the drug substance. If the drug substance is a polymer, an appropriate analytical procedure (eg, size-exclusion chromatography) should be developed to control its content and molecular weight distribution and monitor changes. Depending on the dosage form, the additional tests for vaginal drug products may include viscosity, pH, particle size, dissolution or melting rate, release rate (for controlled-release products), and microbial limits. Although vaginal microbicide products should have a limited bioburden, they are generally not sterile. However, if the manufacture of the vaginal microbicide product includes sterilization, sterility and endotoxin tests should be included in the final drug product specification. For the nonsterile products, microbial limits should be established to assure the absence of specific objectionable microorganisms, and of major importance, testing should be conducted to assure that the product does not support microbial growth. If the product does support microbial growth, it should be reformulated and retested using a minimum amount of an appropriate preservative.
In vitro release testing has been shown to be effective in assessing the product quality and performance over time for certain semisolid dosage forms (eg, creams). However, in vitro release testing should not be used as a surrogate test for in vivo bioavailability or bioequivalence. It is also not appropriate for comparing different semisolid formulations or for comparing similar formulations across manufacturers.
Vaginal microbicides may be used jointly with other methods to prevent HIV and STI transmission (eg, condoms). They may also be employed with other barrier contraceptives. Therefore, it is recommended that the microbicide formulation be evaluated for compatibility with physical barriers such as condoms and diaphragms.
Stability
The stability profile for the drug substance and the drug product should be obtained under long-term and accelerated storage conditions, using analytical methods capable of detecting physical changes and chemical degradation (ie, the analytical procedure should be stability indicating). Initial studies should assure the stability of the drug substance and the drug product for the duration of the proposed clinical trials. Further studies should be performed to support a proposed expiry dating for the commercial product.
Interaction With Genital pH
The site of application of a microbicidal product should be taken into consideration. The pH of the healthy female genital tract is acidic (pH 3.5-4.5). Conversely, the male ejaculate is slightly alkaline with a pH of about 7.5. Thus, the microbicide candidate must be stable over a wide range of pH. In addition, it should not have a negative impact on the acidic pH of the female genital tract.
CONCLUSIONS
Since the first set of recommendations was published in 1996, significant progress has been made in understanding the basic science of HIV transmission. This work has important ramifications for the nonclinical development of microbicides and is reflected in the revised recommendations discussed herein. Under the updated paradigm, there is a greater emphasis on testing the activity of potential microbicides against a wide range of virus types and cell targets in assay systems that reflect the many variables inherent in the process of transmission. The potential risks of local effects have also become clearer, in terms of both the effect of the local environment on the activity of a microbicide and the impact of the microbicide on the local environment. Finally, the value of early, well-integrated formulation research has been recognized and incorporated into the recommended development process. Although it is not possible to give a categorical schema for all products, a general summary of the studies recommended in the nonclinical development of a microbicide candidate is presented in Table 1.
See all tables at end of this report.
The view of the IWGM is that the highest priority needs to be placed on developing a microbicide to prevent or reduce vaginal transmission of HIV in the absence of unacceptable side effects. Consequently, although broad-spectrum activity against other STI pathogens is considered to be a desirable feature, it is not an obligatory one. Similarly, the ability to be formulated for use rectally is desirable, but it is not a requirement. Although the characteristics that are defined as desirable (Table 2) may not be required, the cost of large-scale clinical trials is substantial, and opportunities for large-scale HIV/STI prevention studies are limited. Therefore, the highest priority for clinical development should be given to those products that meet all essential criteria and also have many of the characteristics that are defined as desirable, which include low or no systemic absorption, activity in cell culture and animal model systems against HIV and/or other STI pathogens, high genetic threshold to the development of resistance, good potential for industrial production at low cost, acceptable appearance, odor, consistency, taste, ease of use, and possible rectal application (Table 2). It should be emphasized that even if a candidate microbicide fails to meet some of the listed essential and desirable characteristics, it does not necessarily mean that its development should be discouraged. For example, if a microbicide has activity against vaginal lactobacilli in vitro but possesses other properties that make it a good candidate, its development may be continued. However, data evaluating potential negative effects on the normal flora in the clinical trials should be collected. Similarly, irritation to the rabbit vagina or a high cytotoxic score in vitro may not by itself be a signal to stop development of a particular topical microbicide candidate because their clinical relevance has not been proven. However, it should be reiterated that given the cost and the limited opportunity of large-scale clinical trials, it is prudent to choose the candidate for development carefully.
The information presented in this article is intended to provide general guidance on nonclinical studies that should be conducted to characterize the activity and toxicity of candidate microbicides and to provide supporting data for the initiation of clinical trials. A thorough nonclinical development program is a critical aspect of any microbicide development plan and can be a central contributor to a rapid and successful product development.
ACKNOWLEDGMENTS
The authors thank Drs. Debra Birnkrant, Jim Farrelly, Norman Schmuff, and Teresa Wu of the U.S. Food and Drug Administration for comments and helpful discussions.
The following are members of the IWGM: Consuelo Beck-Sague, Centers for Disease Control and Prevention. Atlanta, GA, USA; Laurent Belec, Hopital European, Paris, France; Beverly Bell, Family Health International, Research Triangle Park, NC, USA; Debra Birnkrant, U.S. Food and Drug Administration, Rockville, MD, USA; Roberta Black, National Institutes of Health, Bethesda, MD, USA; Ward Cates, Family Health International, Research Triangle Park, NC, USA; Lee Claypool, U.S. Agency for International Development, Washington, D.C., USA; Daniel Davis, U.S. Food and Drug Administration, Rockville, MD, USA; Carolyn Deal, National Institutes of Health, Bethesda, MD, USA; Anne Degrand-Guillaud, European Commission, Brussels, Belgium; Ann Duerr, Centers for Disease Control and Prevention, Atlanta, GA, USA; Timothy Farley, World Health Organization, Geneva, Switzerland; Lieve Fransen, European Commission, Brussels, Belgium; Henry Gabelnick, CONRAD Programme, Arlington, VA, USA; Catherine Hankins, UNAIDS, Geneva, Switzerland; Polly Harrison, Alliance for Microbicide Development, Silver Spring, MD, USA; Gerald Jennings, U.S. Agency for International Development, Washington, D.C., USA; Vicky Jespers, Institute of Tropical Medicine, Antwerp, Belgium; Charles Lacey, Imperial College School of Medicine, London, UK; Isaac Malonza, World Health Organization, Geneva, Switzerland; Christine Mauck, CONRAD Programme, Arlington, VA, USA; Charlotte Faty Ndiaye, SWAA, Dakar, Senegal; Andrew Nunn, Medical Research Council, London, UK; Lynn Paxton, Centers for Disease Control and Prevention, Atlanta, GA, USA; Gita Ramjee, Medical Research Council HIV Prevention Research Unit; Durban, South Africa; Patricia Reichelderfer, National Institutes of Health, Bethesda, MD, USA; Zeda Rosenberg, International Partnership for Microbicides, Silver Spring, MD, USA; N.M. Samuel, AIDS Society of India, Chennai, India; Jeffrey Spieler, U.S. Agency for International Development, Washington, D.C., USA; Alan Stone (Chairman), Medical Research Consultant, London, UK; Janet Vail, Program for Appropriate Technology in Health, Seattle, WA, USA; Lut Van Damme, CONRAD Programme, Arlington, VA, USA; Fulvia Veronese, National Institutes of Health, Bethesda, MD, USA; Janneke van de Wijgert, Population Council, New York, NY, USA.
Below is table 1 which had to be reconfigured to fit within the limits of the format of this article. Following recommended studies you will find the purpose for recommending each study and purpose on line 1 refers to recommended study on line 1 and so forth.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
|
|
|