| |
Impact of host genetics on HIV disease progression and treatment: new conflicts on an ancient battleground
|
| |
| |
AIDS: Volume 18(9) 18 June 2004 pp 1231-1240
Nolan, David; Gaudieri, Silvana; John, Mina; Mallal, Simon
From the Centre for Clinical Immunology and Biomedical Statistics, Royal Perth Hospital and Murdoch University, Western Australia.
Introduction
Research into the effects of host genetics on HIV disease susceptibility and progression, and on the efficacy and toxicity of drug treatment, occupies a particular position in the landscape of HIV research. On the one hand, an increased understanding of the role played by host genetic factors in determining the response to HIV infection (for a review see [1]) will undoubtedly contribute to our understanding of the nature of the disease process, and will ultimately assist the development of therapeutic strategies - such as vaccines - that can augment the genetically determined host immune response to HIV infection. In a limited number of cases a favourable host genetic profile will provide protection from HIV infection, or will confer 'long-term non-progressor' status following infection, allowing durable control of HIV-associated disease progression without the requirement for drug therapy. On the other hand, the relevance of these genetic factors to the routine clinical management of HIV-infected patients is often perceived as being marginal compared with the cost and difficulty associated with undertaking genetic testing, so that the assessment of host genetic factors is not incorporated into standard guidelines for HIV management [2,3]. Moreover, whilst genetic factors may significantly alter the rate of disease progression at an individual level, clinical decisions are currently based on defined endpoints such as CD4 T-cell number, HIV viral load, rate of CD4 T-cell decline and the clinical expression of AIDS- defining illnesses, irrespective of the host factors that may determine these values [2,3].
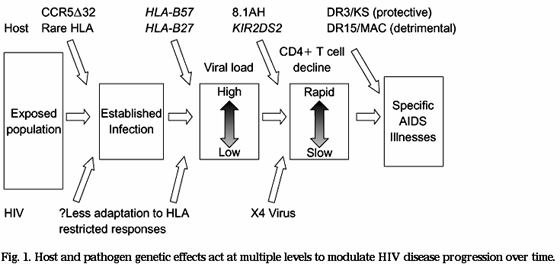
Several reviews are available that enumerate those host genetic factors that have a demonstrable effect on the course of HIV infection, including a recent comprehensive assessment of this field of research [1]. Here we will attempt to provide a broad conceptual framework for considering the impact of host genetic diversity on HIV/AIDS; one that argues that host genetic variation is an important factor in the response to HIV for every infected individual, and within every infected population; not just for those few individuals who carry genetic variants that have powerful protective (or deleterious) effects on HIV disease progression [4]. This review incorporates an evolutionary perspective that recognizes the ongoing dynamic interplay between host and viral genetic diversity at the population level, providing the driving force for the selection of favourable host genetic profiles as well as for viral mutations that are best adapted to these host effects. We also present a clinical perspective that considers analogies between 'endogenous' antiviral effects conferred by a broad spectrum of host genetic factors, and the effects of drug therapy. This model provides for a broad conception of the role of host factors in determining the overall progression of HIV disease, both before and after the introduction of antiretroviral therapy (Fig. 1).
HOST AND PATHOGEN GENETIC EFFECTS ACT AT MULTIPLE LEVELS TO MODULATE HIV DISEASE PROGRESSION OVER TIME
|
|
| |
| |
 |
|
| |
| |
An evolutionary perspective: host-pathogen interactions at the genetic level
One interesting by-line in the story of the human genome project has been that our DNA is littered with the remnants of previous encounters with retroviruses. It is estimated that approximately 8% of human genetic sequence is derived directly from retroviral elements, and that subsequent transposition and recombination events have amplified this proportion to above 40% [5]. Moreover, the more recent human endogenous retroviruses have been acquired in the period since human evolution diverged from our nearest primate ancestors [6]. We have therefore encountered retroviral pathogens throughout our evolutionary history, and these infectious agents are likely to have shaped the evolution of critical aspects of our immunological response to viral infection. Recent evidence points to the purifying pressure that may be exerted by pathogenic viruses on host genetic diversity, suggesting that the selection of a limited number of major histocompatibility complex (MHC) alleles able to direct a strong protective immunological response against both SIV and the closely related human lentivirus HIV-1 has been a feature of the successful adaptation made by chimpanzees to SIV [7]. Evolutionary history is punctuated by these bottleneck events in which diversity is sacrificed in order to ensure the survival of populations in the face of epidemic or endemic pathogens, and it is these events that are likely to have determined the non-random distribution of MHC variants in different ethnically and geographically separated groups [8-11]. It is also interesting to observe, however, that the incorporation of retroelements into the human genome creates sites of genetic instability that have contributed to large-scale genetic duplication and recombination events - including within the MHC region - thus producing quantum evolutionary changes and the development of new genetic loci critical for immune function [12].
If we consider populations both past and present with no access to effective treatment for HIV infection, the only 'weapons' available to combat the spread of HIV are host genetic diversity (at a given time) and host genetic adaptation (developing over generations under the selective pressure of endemic HIV infection). Given that HIV is capable of rapid genetic adaptation to the host immune response, producing multiple generations of virions every day, it would be anticipated that the pathogen has a marked advantage in this conflict. However, in the human host's favour, there is a remarkable degree of diversity in key regions of the genome that encode for proteins that shape the immune response, so that adaptation to one individual does not provide a particularly strong advantage when infection is passed on to the next host. It is this 'strength in diversity' that defines the host population's most significant natural advantage; one that needs to be exploited in the development of therapeutic strategies such as vaccines [13].
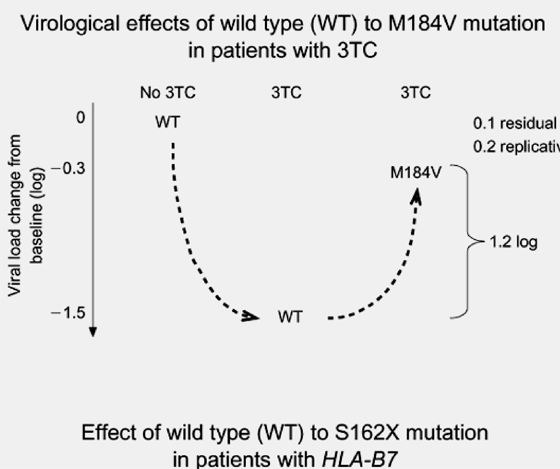
Can we extrapolate this evolutionary perspective to the present era in any useful way? After more than 20 years of epidemic HIV infection (i.e., approximately one human generation), it is probably too early to see if human HLA diversity is being affected by this selective pressure. However, this time scale is more than ample for the generation of viral diversity in response to the genetically encoded human immune system. In this context, the ability of autologous HIV to make the appropriate mutational adaptations to HLA-restricted immune response has been demonstrated at the population level, where it predicts pre-treatment viral load setpoint [14]. This capacity for adaptation may be determined by: (i) the genetic barrier to escape (i.e., the number of mutational steps required to induce escape at a specific viral amino acid residue); (ii) costs to viral replicative fitness associated with escape mutations; (iii) the breadth of the HLA-restricted immune response to viral antigens (i.e., the number of epitopes recognized by a specific HLA allele); and (iv) the residual cytotoxic T lymphocyte (CTL) response present following escape mutation (see Fig. 2). We can therefore see that there is a dynamic relationship between the host and the infecting virus that is played out at the (host) individual level, as the virus seeks to evade the immune responses of the host in which it finds itself. This also translates to the population level, as HIV is passed from one individual to another through a population. It is therefore worth bearing in mind when considering the impact of specific host genetic factors that HIV represents a constantly shifting target for the host immune response.
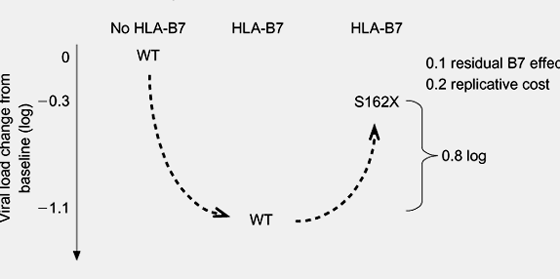
Fig. 2. Analogies between the effects of drug resistance mutations and CTL escape mutations on viral load. (a) In the presence of a selected antiretroviral drug [in this case, lamivudine (3TC)], there is a reduction of viral load associated with effective therapy (1.5 log10copies/ml) in this hypothetical scenario. The subsequent development of a M184V resistance mutation under the selective pressure provided by 3TC is associated with a rebound of viral load due to drug resistance. However, the increase in viral load is offset in this case by a reduction of viral fitness, and some residual effect of 3TC on the mutated virus, so that the viral load does not return to baseline levels while 3TC is continued (net loss, 1.2 log10copies/ml). (b) Similarly, the CTL response to HIV-1 associated with HLA-B7 results in a reduction of viral load of approximately 1.1 log10copies/ml. However, the development of a S162X CTL escape mutation within the HIV reverse transcriptase sequence is associated with loss of HLA-B7-specific immune recognition at this site, and an increase in viral load that is again offset by reduced viral fitness and residual effects of HLA-B7 on mutated virus.
|
|
| |
| |
 |
|
| |
| |
THIS WAS CUT-OFF FROM GRAPH
0.1 residual 3TC effect
0.2 replicative cost
|
|
| |
| |
 |
|
| |
| |
Host genetic factors and the natural history of HIV disease progression
The host immune response to HIV infection is shaped to a large extent by an individual's repertoire of antigen recognition molecules encoded within the MHC on chromosome 6. This region of the human genome is characterized by intense polymorphism, with over 1600 individual alleles currently known at the major HLA loci (Class I, HLA-A, HLA-B, HLA-C; Class II, HLA-DR). Hence, the inheritance of HLA alleles as well as polymorphic immunoregulatory genes within the central MHC region that are in strong linkage disequilibrium with them (e.g., cytokines such as tumour necrosis factor-a, chaperone proteins such as heat shock proteins), profoundly influences the capacity to recognize and destroy pathogens that have managed to breach the defences provided by protective mucosal barriers and innate immune responses. The adaptive immune response also uses auxiliary systems of antigen recognition that are also highly polymorphic, such as the killer inhibitory receptor (KIR) gene cluster on chromosome 19 [18-21] which is involved in modulating immune responses through recognition of 'missing self' (identifying the absence of Class I MHC surface expression) as well as of specific ligand epitopes present on HLA-C and HLA-B alleles [18-21]. Polymorphic variation in chemokine receptor expression is also an important source of host defence against HIV [1], which is to some degree countered by the virus' ability to utilize alternative coreceptors for cellular entry. Finally, there are a number of novel mechanisms used by the human host to neutralize the metabolic and transcriptional machinery employed by HIV for ongoing replication, which may be exemplified by the ability of the APOBEC3G gene product to induce hypermutation in the HIV gene sequence by interfering with pyrimidine nucleotide metabolism [22-24]. These non-antigen specific mechanisms are not subject to as much
genetic polymorphism, and have in general provoked HIV to 'solve' the problems they pose (for example, the ability of the Vif regulatory protein to counter the antiviral effects of the APOBEC3G product [24-26]).
Impact of MHC alleles and haplotypes on HIV disease progression
Given that the immune response to viral antigens mediated by cytotoxic CD8 T cells is dependent on the presence of Class I MHC molecules (e.g., HLA-A and HLA-B), and that the recognition of peptide motifs by this system is highly specific for a given HLA allele, it is perhaps not surprising that the inheritance of certain Class I alleles has been associated with significantly more (or in some cases, less) effective immunological control of HIV infection compared with others [1,27]. A number of studies have identified effects of HLA alleles that are favourable in some cases (e.g., HLA-B57, HLA-B27) and unfavourable in others (e.g., HLA-B35 and HLA-B22 subtypes) [1,27-36], and have demonstrated that the combined effects of HLA alleles are predictive of long-term non-progressor status [35] and viral load set-point in the pre-treatment phase [30-32]. HLA-B18 has also been found to predict slower progression of HIV disease [31], and has also been linked to protection from acquiring HIV infection in two studies involving multiply exposed sex workers in Thailand [37] and Kenya [38]. As previously mentioned, these dominant allelic associations represent extreme examples of the dynamic relationship between HLA-restricted antigen recognition and the capacity for HIV to evade immune surveillance through mutation [14]. At one end of this spectrum are the powerful protective alleles such as HLA-B57 and HLA-B27, whilst at the other end of the spectrum are alleles that may recognize epitopes with few constraints to generating successful viral escape mutations. In each case, it is the effect on viral load setpoint that is primarily responsible for genetic associations with altered disease progression. As presented in Fig. 2, the factors outlined above that determine the antiviral efficacy of specific HLA alleles are analogous to those determining antiretroviral drug efficacy. Hence, concepts that are familiar to clinicians who manage HIV therapy may also be applied to the 'endogenous drugs' that are represented by inherited HLA alleles.
Modulating HIV disease progression at multiple levels
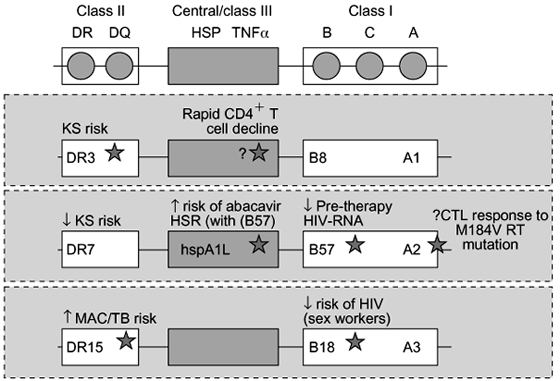
In certain cases, the influence of HLA alleles on disease progression appears to extend beyond a direct effect on HIV viral load (Fig. 1). For example, individuals who carry HLA-B8 as well as other genetic markers within the MHC region that are strongly linked to this allele (denoted the 8.1 ancestral haplotype) experience a more rapid CD4 T-cell decline [39], particularly early in the course of disease (from seroconversion to CD4 < 20%; Fig. 3) [40]. Further genetic mapping of this association has shed some light on the CD4 T cell-depleting effect of this haplotype, indicating that this propensity is associated with unknown polymorphisms present within the central MHC region, rather than with HLA-B8 itself [17]. Hence, it is plausible that immunoregulatory gene products encoded in this region determine the efficiency of immune-mediated CD4 T-cell destruction in response to a given HIV burden. This concept is supported by observations that a relative deficiency of type-1 cytokine production, characteristically seen with the HLA-B8/-DR3- associated MHC haplotype [41], is associated with increased susceptibility to activation-induced CD4 T-cell death [42].
Fig. 3. Effects of HLA alleles on HIV disease progression may reflect associations with strongly linked genes within the MHC region. Examples are provided of MHC haplotypes and their associations with HIV disease progression, noting that HLA alleles and linked immunoregulatory genes within a common MHC haplotype may have synergistic or competing effects on overall progression of HIV/AIDS. KS, Kaposi's sarcoma; TNF-a, tumour necrosis factor-alpha; HSP, heat shock protein; HSR, hypersensitivity syndrome; MAC/TB, Mycobacterium avium complex/tuberculosis.
|
|
| |
| |
 |
|
| |
| |
Looking beyond the direct consequences of HIV infection (i.e., HIV viral load and CD4 T-cell decline), genetic factors also appear to independently modulate susceptibility to infections caused by opportunistic intracellular pathogens. For example, risk of developing HHV-8-associated Kaposi's sarcoma has been associated with HLA-DR5 in both 'classical' [43,44] forms of the disease as well as in HIV-associated Kaposi's sarcoma (along with HLA-DR1) [45-51] in Caucasian (but not African [52]) populations, whilst HLA-DR3 (a component of the HLA-B8-associated ancestral MHC haplotype) is protective [50,51]. In the case of the common Caucasian MHC haplotype that includes HLA-A1/ -B8/-DR3 alleles, these data indicate that individuals carrying this haplotype experience more rapid CD4 T-cell decline and are generally at increased risk of opportunistic infections; but at the same remain at low risk of developing Kaposi's sarcoma [50,51]. Susceptibility to disseminated Mycobacterium avium complex infection has also been linked to Class II MHC region, in this case to the closely linked HLA-DRB1*1501 and -DQ*0602 alleles previously associated with risk of tuberculosis and leprosy in populations where these diseases are prevalent [53,54]. Immune restoration disease (IRD) associated with aberrant inflammatory responses to pathogens following the introduction of antiretroviral therapy have also been associated with HLA alleles, with significant associations noted between the HLA-B44/-DR4 MHC haplotype and cytomegalovirus-related IRD [55].
These strong genetic risk factors indicate that susceptibility to specific opportunistic diseases is not simply a product of the severity of immunosuppresion, but is also modulated by the genetically determined immune response to the relevant pathogen; again suggesting that 'progression to AIDS' is a term that encompasses a broad range of pathological events. Additionally, HLA alleles expressed in the context of broader MHC haplotypes may have complex associations with HIV/AIDS progression, as outlined in Fig. 3.
Impact of chemokine receptor polymorphism
The disease-modifying effect of a 32-base pair deletion in the CCR5 receptor gene (CCR5D32) present in around 10% of Caucasians, and the significant protection against HIV-1 infection afforded by homozygosity at this allele, represents the prototypic association between a host genetic factor and altered HIV disease progression (reviewed in [1,56]). Interestingly, this genetic variant may have arisen in Northern European populations comparatively recently [57] under selective pressure provided by epidemic viral pathogens such as smallpox [58] or variola [59].
With increasing knowledge of the haplotype structure of the chemokine receptor genes, and the role of chemokine ligands such as MIP-1a, MIP-1b and Rantes in modulating chemokine receptor function, it has become apparent that numerous functional polymorphisms exist in these genetic systems; extending the 'breadth' of effects of chemokine receptor variation far beyond a model with CCR5D32) as a single important loss-of-function variant. This topic has been elegantly reviewed elsewhere [1,56], and the issue of genetic variation in these interacting systems has been addressed in a large multinational study that also points out that effects of these host genetic factors on HIV/AIDS differ according to the population studied [61]. Hence, the overall impact of chemokine receptor polymorphism and chemokine receptor ligand polymorphism may be considered in a similar manner to the MHC system, in that genetic variation is relevant to the response to HIV-1 infection in most if not all individuals, whilst dominant effects on disease progression (e.g., CCR5D32) are relatively uncommon.
The impact of chemokine receptor genotype may also provide some insights regarding the interdependent nature of host genetic effects on disease progression. First, the dominant genetic effect of the CCR5D32 receptor mutation can provide a significant barrier to successful HIV infection at an individual level, providing significant associations with delayed disease progression and long-term non-progressor status [1,56]. However, the predictive value of CCR5D32 on disease progression appears to be dependent on: (i) disease stage, with evidence for early effects that are subsequently lost as duration of HIV infection increases [62-64]; and (ii) presence of other host genetic factors, particularly HLA, that combine with CCR5D32 to determine 'non-progressor' or 'progressor' status [65,66]. This is not to downplay the impact of the CCR5D32 mutation or other protective chemokine receptor mutations that can certainly delay disease progression to an extent that is clinically relevant [1], but to place these genetic factors within an overall 'landscape' of selective pressure in untreated individuals (Fig. 4). For example, CCR5D32 heterozygosity is not in itself sufficient to confer non-progressor status [65,66], nor is carriage of the most potent protective HLA allele, HLA-B*5701 [67]. However, each of these host genetic factors appears to contribute to a more broadly defined selective pressure provided by multiple genetically determined factors [29-31,34] (Fig. 4). In the case of chemokine receptor gene polymorphisms, by providing another source of selective pressure (in this case, against the emergence of CCR5-dependent M-tropic viral strains) [68], these host genetic effects contribute to the overall immunological and/or drug pressure against which HIV must adapt to optimize viral fitness. The CCR5D32 mutation may also augment the magnitude and durability of virological responses to HIV therapy [69-71] consistent with an ongoing contribution to overall virological control during treatment (Fig. 5), although this association has not been consistently observed [72,73].
Fig. 4. Multiple potential selective pressures act on HIV. In this context, effects of selective pressure from multiple sources may contribute to overall ability to effectively control viral load and HIV-1 disease progression, even in the presence of antiretroviral drug therapy.
|
|
| |
| |
 |
|
| |
| |
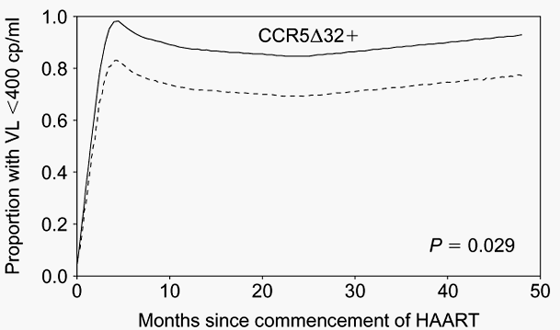
Fig. 5. Influence of CCR5D32 mutation on virological response to antiretroviral therapy in the Western Australian HIV cohort. In this analysis of profiles of post-treatment viral response proportions among antiretroviral therapy-naive white males commencing HAART, presence of the CCR5D32 mutation was associated with an improved virological response to antiretroviral therapy early in the treatment course and over the subsequent 50 months (P = 0.029). It is notable that the baseline viral load of the CCR5D32-positive group (4.67 log10 copies/ml, n = 19) was lower than the CCR5D32- -negative group (5.12 log10 copies/ml, n = 78) (P = 0.01), and that after adjustment for baseline viral load the difference in virological response to treatment between the groups was no longer significant (P = 0.11). These data suggest that the effect of CCR5D32 on levels of viraemia continues to have an impact during antiretroviral therapy, consistent with the schema provided in Fig. 4.
|
|
| |
| |
 |
|
| |
| |
Potential role of KIR genotypes and haplotypes
The KIR complex on chromosome 19 is a highly polymorphic region of the genome which encodes receptors that are widely expressed on natural killer cells. Knowledge of the genomic structure of this region is still incomplete, and the natural ligands for a number of KIR gene products remain unknown. However, cognate interactions between killer immunoglobulin-like receptors and their ligands may inhibit alloreactivity and cytotoxicity, but may also activate cell-mediated immune responses depending on the KIR gene involved [18-21].
The involvement of the KIR complex in modulating HIV disease progression has been a subject of increasing attention in recent times. For example, a known KIR recognition motif that is common to a number of HLA-B alleles (denoted Bw4) has been associated with slower HIV disease progression [74], with further data suggesting that the interaction of the Bw4 motif and a specific activating KIR (KIR3DS1) may explain this association [75]. However, strong linkage disequilibrium within the KIR complex makes it difficult to assign effects to individual KIR genes within a haplotype. In recent studies we have identified associations between rapid CD4 T-cell decline and a KIR haplotype that contains KIR2SD2 and a predominance of activatory KIR alleles [76]. These data suggest that the effect of KIR genotype on HIV disease progression may modify the susceptibility of CD4 T cells to undergo lysis, rather than affecting viral load set point, possibly reflecting the ability of immune cells expressing KIR to recognize and destroy HIV-infected CD4 T cells through recognition of both 'positive' signals (e.g., epitopes expressed on HLA-C and HLA-B alleles) and 'negative' signals (e.g., reduced Class I MHC expression - `missing self') induced by regulatory viral proteins such as nef [77]. In this context, the genetically determined balance between inhibitory and activatory KIR alleles may play an important role in shaping the immunological response, along with the inheritance of HLA alleles with which KIR alleles interact.
Host genetic factors and the response to HIV therapy
Here the major focus of research has been on potential associations between drug hypersensitivity reactions and HLA alleles. This effort has borne fruit in the case of abacavir hypersensitivity, where two independent studies have demonstrated strong associations between HLA-B*5701 and susceptibility to this syndrome [78,79]. The predictive value of genetic testing may be further improved with the incorporation of a central MHC polymorphism (HSPA1L within the heat shock protein gene cluster) as well as HLA-B*5701, which in one study increased the positive predictive value of testing to > 90% and the negative predictive value to > 99% [80]. This is certainly one area of research into host genetic effects that may have direct implications for clinical practice, as the avoidance of this hypersensitivity syndrome through prospective genetic testing may significantly reduce the burden of toxicity- associated morbidity (and mortality) associated with the use of abacavir [80].
It is also possible that susceptibility to nevirapine hypersensitivity - manifesting as potentially life-threatening hepatotoxicity with or without rash - is also conferred by genetic factors, as this syndrome is similar to abacavir hypersensitivity in that susceptible individuals develop symptoms after short-term exposure (including HIV-negative patients receiving nevirapine for post-exposure prophlaxis) [81-83]. The protective effect of lower CD4 T-cell counts in the case of nevirapine hypersensitivity [84], however, suggests that a genetic association is more likely to involve Class II MHC alleles such as HLA-DR, as indeed is the case for most of the described HLA-drug associations.
Another aspect of HLA-drug associations that has not yet received much attention is the potential for HLA alleles to influence the efficacy of drug therapy. This possibility arises from the notion that mutations selected by drug therapy may also be sites where selective pressure is provided by HLA alleles. Whilst the resistance mutations associated with specific antiretroviral drugs are generally distinct from 'natural' escape mutations described above, interactions may nevertheless exist, as the selective pressure of drug therapy provides a new impetus for site-specific mutations. This may result in: (i) the generation of novel epitopes for HLA-restricted immune recognition, as has been demonstrated for the M184V reverse transcriptase mutation which can act as a neoepitope for HLA-A2 [85]; and (ii) facilitation (or impairment) of the capacity for escape mutations to develop at the site of a drug resistance mutation, or at more distant sites through effects on conformational structure and/or charge of peptide sequences. For example, a protease inhibitor resistance mutation (V82A) that occurs within a high-avidity HLA-A2-restricted epitope (residues 76-84) leads to abrogation of immune recognition at this site, so that the protease inhibitor-selected V82A mutation acts as a protease inhibitor and a CTL escape mutant in HLA-A2-positive individuals [86]. More work is required in this area, but it is conceivable that altered risk of developing drug resistance mutations associated with certain HLA alleles may prove to be sufficiently strong in some cases to influence the choice of antiretroviral regimen in the future.
Combining host and drug antiviral effects: clinical utility of host genetic factors?
At a fundamental level, it may be that drug therapy for HIV is more effective when there is also a contribution from the host immune response towards the suppression of viral load. Evidence supporting this proposition has been provided by studies demonstrating that CTL responses to HIV remains relevant during HIV treatment [87-92], particularly in the presence of incompletely suppressive treatment associated with detectable viraemia [89,90], and that this immune response contributes to the virological response to therapy [89,90]. Importantly, the effectiveness of CTL responses during therapy is predicted by those measured prior to commencing treatment [87,88], an observation that may explain why high viral load at baseline [93] and early in the course of treatment with HAART regimens [94,95] is associated with earlier virological failure [96] and higher rates of HIV-associated mortality [93].
It may therefore be possible to employ the same pragmatic approach to 'managing' host genetic effects that is already familiar to prescribers of antiretroviral therapy. That is, the level of viraemia in the pre-treatment stage provides a surrogate measure of the overall selective pressure provided by host genetic factors, and the degree to which the autologous virus has been able to adapt to this host immune response. For individuals with residual immunological control of viraemia, it may be argued that the viral load should be considered along with CD4 T-cell count when deciding when to initiate antiretroviral therapy, and which regimens to use. Delaying therapy until the viral load is very high (i.e., > 100 000 copies HIV RNA/ml) is likely to place a greater reliance on the efficacy of drug therapy to control viraemia, so that optimally effective first-line therapy is paramount in these individuals. On the other hand, initiating treatment while virological control is still present may allow for improved long-term outcomes by combining 'endogenous drugs' with antiretroviral therapy to effectively combat HIV (Fig. 4). Preserving the activity of endogenous antiviral factors for as long as possible may also have particular importance in compartments such as the central nervous system that may receive limited benefit from antiretroviral treatment [97].
Conclusions
It is argued here that our conception of host genetic factors as determinants of HIV disease progression needs to extend beyond those few examples where allelic variation at a single genetic locus confers a significant beneficial or harmful effect, to see that genetic variation operates at multiple levels to modulate disease progression for each individual and across populations. Recognizing that genetically determined factors operate in important ways in all infected individuals to modulate the immunological response to HIV infection, and that this variable host response then influences the evolution and diversity of the viral genome, allows for more powerful approaches to investigate both the immunological and virological aspects of HIV disease at the population level. Acknowledging the important contribution of host factors in controlling HIV infection throughout the course of the disease process - including the treatment phase - may also allow for increasingly optimized approaches to long-term HIV management.
This knowledge can then be utilized in a number of ways. From a research perspective, an understanding of the inherently complex and dynamic nature of host-pathogen interactions can shape the development of appropriate research methods. For example, utilizing 'progression to AIDS' as a study endpoint may encompass diverse effects including: susceptibility to HIV infection following exposure, HIV viral load setpoint, rate of CD4 T-cell decline, and susceptibility to specific AIDS-defining illnesses. Analysing these outcomes as independent and potentially interactive variables, however, may shed light on the underlying pathophysiology and help to define potential targets for therapeutics. Perhaps the most immediate and practical application of such knowledge is in vaccine development, a long sought after advance in HIV prevention, where a detailed knowledge of dynamic host-viral interactions, and how these interactions at the individual level then play out at the population level, is paramount for success. In this way, the advantages provided by the intensity of our host genetic diversity can be exploited to overcome the problems associated with HIV diversity; a concept that may be illustrated by a quotation attributed to the American president Abraham Lincoln:
`You can fool some of the people all of the time, and all of the people some of the time; but you can't fool all of the people all of the time.'
References1. Tang J, Kaslow R. The impact of host genetics on HIV infection and disease progression in the era of highly active antiretroviral therapy. AIDS 2003, 17(Suppl 1):S51-S60.
2. British HIV Association guidelines available at http://www.bhiva.org
3. United States Department of Health and Human Servises (DHHS) guidelines available at http://aidsinfo.nih.gov
4. Carrington M, Nelson G, O'Brien SJ. Considering genetic profiles in functional studies of immune responsiveness to HIV-1. Immunol Lett 2001, 79:131-140.
5. Griffiths DJ. Endogenous retroviruses in the human genome sequence. Genome Biol 2001, 2:1017.1-1017.5.
6. Tristem M. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J Virol 2000, 74:3715-3730.
7. de Groot NG, Otting N, Doxiadis GG, Balla-Jhagjhoorsingh SS, Heeney JL, van Rood JJ, et al. Evidence for an ancient selective sweep in the MHC class I gene repertoire of chimpanzees. Proc Natl Acad Sci USA 2002, 99:11748-11753.
8. Van Blerkom LM. Role of viruses in human evolution. Am J Phys Anthropol 2003, 37 (Suppl):14-46.
9. Klein J, Gutknecht J, Fischer N. The major histocompatibility complex and human evolution. Trends Genet 1990, 6:7-11.
10. Meyer D, Thomson G. How selection shapes variation of the human major histocompatibility complex: a review. Ann Hum Genet 2001, 65:1-26.
11. Apanius V, Penn D, Slev PR, Ruff LR, Potts WK The nature of selection on the major histocompatibility complex. Crit Rev Immunol 1997, 17:179-224.
12. Kulski JK, Gaudieri S, Bellgard M, Balmer L, Giles K, Inoko H, Dawkins RL. The evolution of MHC diversity by segmental duplication and transposition of retroelements. J Mol Evol 1997, 45:599-609.
13. Mehra NK, Kaur G, Jaini R. Genetic diversity in the Human Major Histocompatibility Complex: lessons for vaccination approaches to HIV infection. Community Genet 2002, 5:162-166.
14. Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 2002, 296:1439-1443.
15. Klein J, Sato A. The HLA system. First of two parts. N Engl J Med 2000, 343:702-709.
16. Klein J, Sato A. The HLA system. Second of two parts. N Engl J Med 2000, 343:782-786.
17. Price P, Witt C, Allcock R, Sayer D, Garlepp M, Kok CC, et al. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev 1999, 167:257-274.
18. Martin AM, Freitas EM, Witt CS, Christiansen FT. The genomic organization and evolution of the natural killer immunoglobulin-like receptor (KIR) gene cluster. Immunogenetics 2000, 51: 268-280.
19. Hsu KC, Chida S, Geraghty DE, Dupont B. The killer cell immunoglobulin-like receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism. Immunol Rev 2002, 190:40-52.
20. Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol 2002, 20:217-251.
21. Long EO, Burshtyn DN, Clark WP, Peruzzi M, Rajagopalan S, Rojo S, Wagtmann N, Winter CC. Killer cell inhibitory receptors: diversity, specificity, and function. Immunol Rev 1997, 155:135-144.
22. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424:99-103.
23. Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424:94-98.
24. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418:646-650.
25. Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 2003, 9:1404-1407.
26. Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302:1056-1060.
27. O'Brien SJ, Gao X, Carrington M. HLA and AIDS: a cautionary tale. Trends Mol Med 2001, 7:379-381.
28. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 2004; 10:282-289.
29. Altfeld M, Addo MM, Rosenberg ES, Hecht FM, Lee PK, Vogel M, et al. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 2003, 17: 2581-2591.
30. Tang J, Wilson CM, Meleth S, Myracle A, Lobashevsky E, Mulligan MJ, et al. Host genetic profiles predict virological and immunological control of HIV-1 infection in adolescents. AIDS 2002, 16:2275-2284. 31. Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med 1996, 2:405-411.
32. Ioannidis JP, Goedert JJ, McQueen PG, Enger C, Kaslow RA. Comparison of viral load and human leukocyte antigen statistical and neural network predictive models for the rate of HIV-1 disease progression across two cohorts of homosexual men. J Acquir Immune Defic Syndr Hum Retrovirol 1999, 20: 129-136. 33. Dorak MT, Tang J, Tang S, Penman-Aguilar A, Coutinho RA, Goedert JJ, et al. Influence of human leukocyte antigen-B22 alleles on the course of human immunodeficiency virus type 1 infection in 3 cohorts of white men. J Infect Dis 2003, 188: 856-863.
34. Flores-Villanueva PO, Hendel H, Caillat-Zucman S, Rappaport J, Burgos-Tiburcio A, Bertin-Maghit S, et al. Associations of MHC ancestral haplotypes with resistance/susceptibility to AIDS disease development. J Immunol 2003, 170:1925-1929.
35. Magierowska M, Theodorou I, Debre P, Sanson F, Autran B, Riviere Y, et al. Combined genotypes of CCR5, CCR2, SDF1, and HLA genes can predict the long-term nonprogressor status in human immunodeficiency virus-1-infected individuals. Blood 1999, 93:936-941.
36. Hendel H, Caillat-Zucman S, Lebuanec H, Carrington M, O'Brien S, Andrieu JM, et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J Immunol 1999, 162:6942-6946.
37. Beyrer C, Artenstein AW, Rugpao S, Stephens H, VanCott TC, Robb ML, et al. Epidemiologic and biologic characterization of a cohort of human immunodeficiency virus type 1 highly exposed, persistently seronegative female sex workers in northern Thailand. Chiang Mai HEPS Working Group. J Infect Dis 1999, 179:59-67.
38. Rowland-Jones SL, Dong T, Fowke KR, Kimani J, Krausa P, Newell H, et al. Cytotoxic T cell responses to multiple conserved HIV epitopes in HIV-resistant prostitutes in Nairobi. J Clin Invest 1998, 102:1758-1765.
39. Kaslow RA, Duquesnoy R, VanRaden M, Kingsley L, Marrari M, Friedman H, et al. A1, Cw7, B8, DR3 HLA antigen combination associated with rapid decline of T-helper lymphocytes in HIV-1 infection. A report from the Multicenter AIDS Cohort Study. Lancet 1990, 335:927-930.
40. Mallal S, Cameron PU, French MA, Dawkins RL. MHC genes and HIV infection. Lancet 1990, 335:1591-1592. 41. Candore G, Romano GC, D'Anna C, Di Lorenzo G, Gervasi F, Lio D, et al. Biological basis of the HLA-B8,DR3-associated progression of acquired immune deficiency syndrome. Pathobiology 1998, 66:33-37.
42. Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol 2003, 84:1649-1661.
43. Kaloterakis A, Papasteriades C, Filiotou A, Economidou J, Hadjiyannis S, Stratigos J. HLA in familial and nonfamilial Mediterranean Kaposi's sarcoma in Greece. Tissue Antigens 1995, 45:117-119.
44. Contu L, Cerimele D, Pintus A, Cottoni F, La Nasa G. HLA and Kaposi's sarcoma in Sardinia. Tissue Antigens 1984, 23: 240-245.
45. Pollack MS, Safai B, Myskowski PL, Gold JW, Pandey J, Dupont B. Frequencies of HLA and Gm immunogenetic markers in Kaposi's sarcoma. Tissue Antigens 1983, 21:1-8.
46. Scorza Smeraldi R, Fabio G, Lazzarin A, Eisera NB, Moroni M, Zanussi C. HLA-associated susceptibility to acquired immunodeficiency syndrome in Italian patients with human-immunodeficiency-virus infection. Lancet 1986, 2:1187-1189.
47. Prince HE, Schroff RW, Ayoub G, Han S, Gottlieb MS, Fahey JL. HLA studies in acquired immune deficiency syndrome patients with Kaposi's sarcoma. J Clin Immunol 1984, 4:242-245.
48. Jeannet M, Sztajzel R, Carpentier N, Hirschel B, Tiercy JM. HLA antigens are risk factors for development of AIDS. J Acquir Immune Defic Syndr 1989, 2:28-32.
49. Pollack MS, Gold J, Metroka CE, Safai B, Dupont B. HLA-A,B,C and DR antigen frequencies in acquired immunodeficiency syndrome (AIDS) patients with opportunistic infections. Hum Immunol 1984, 11:99-103.
50. Klein MR, Keet IP, D'Amaro J, Bende RJ, Hekman A, Mesman B, et al. Associations between HLA frequencies and pathogenic features of human immunodeficiency virus type 1 infection in seroconverters from the Amsterdam cohort of homosexual men. J Infect Dis 1994, 169:1244-1249.
51. Mann DL, Murray C, O'Donnell M, Blattner WA, Goedert JJ. HLA antigen frequencies in HIV-1-related Kaposi's sarcoma. J Acquir Immune Defic Syndr 1990, 3 (Suppl 1):S51-S5.
52. Melbye M, Kestens L, Biggar RJ, Schreuder GM, Gigase PL. HLA studies of endemic African Kaposi's sarcoma patients and matched controls: no association with HLA-DR5. Int J Cancer 1987 Feb 15;39(2):182-4.
53. LeBlanc SB, Naik EG, Jacobson L, Kaslow RA. Association of DRB1*1501 with disseminated Mycobacterium avium complex infection in North American AIDS patients. Tissue Antigens 2000, 55:17-23.
54. Naik E, LeBlanc S, Tang J, Jacobson LP, Kaslow RA. The complexity of HLA class II (DRB1, DQB1, DM) associations with disseminated Mycobacterium avium complex infection among HIV-1-seropositive whites. J Acquir Immune Defic Syndr 2003, 33:140-145.
55. Price P, Keane NM, Stone SF, Cheong KY, French MA. MHC haplotypes affect the expression of opportunistic infections in HIV patients. Hum Immunol 2001, 62:157-164.
56. Carrington M, Dean M, Martin MP, O'Brien SJ. Genetics of HIV-1 infection: chemokine receptor CCR5 polymorphism and its consequences. Hum Mol Genet 1999, 8:1939-1945.
57. Libert F, Cochaux P, Beckman G, Samson M, Aksenova M, Cao A, et al. The deltaccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe. Hum Mol Genet 1998, 7:399-406.
58. Galvani AP, Slatkin M. Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV- resistance allele. Proc Natl Acad Sci USA 2003, 100: 15276-15279.
59. Lucotte G. Frequencies of 32 base pair deletion of the (Delta 32) allele of the CCR5 HIV-1 co-receptor gene in Caucasians: a comparative analysis. Infect Genet Evol. 2002, 1:201-205.
60. Mummidi S, Ahuja SS, Gonzalez E, Anderson SA, Santiago EN, Stephan KT, et al. Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression. Nat Med 1998, 7:786-793.
61. Gonzalez E, Dhanda R, Bamshad M, Mummidi S, Geevarghese R, Catano G, et al. Global survey of genetic variation in CCR5, RANTES, and MIP-1alpha: impact on the epidemiology of the HIV-1 pandemic. Proc Natl Acad Sci USA 2001, 98:5199-5204.
62. Hendel H, Henon N, Lebuanec H, Lachgar A, Poncelet H, Caillat-Zucman S, et al. Distinctive effects of CCR5, CCR2, and SDF1 genetic polymorphisms in AIDS progression. J Acquir Immune Defic Syndr Hum Retrovirol 1998, 19:381-386.
63. Tang J, Shelton B, Makhatadze NJ, Zhang Y, Schaen M, Louie LG, et al. Distribution of chemokine receptor CCR2 and CCR5 genotypes and their relative contribution to human immunodeficiency virus type 1 (HIV-1) seroconversion, early HIV-1 RNA concentration in plasma, and later disease progression. J Virol 2002, 76:662-672.
64. Mulherin SA, O'Brien TR, Ioannidis JP, Goedert JJ, Buchbinder SP, Coutinho RA, et al. Effects of CCR5-Delta32 and CCR2-64I alleles on HIV-1 disease progression: the protection varies with duration of infection. AIDS 2003, 17:377-387.
65. Morawetz RA, Rizzardi GP, Glauser D, Rutschmann O, Hirschel B, Perrin L, et al. Genetic polymorphism of CCR5 gene and HIV disease: the heterozygous (CCR5/delta ccr5) genotype is neither essential nor sufficient for protection against disease progression. Swiss HIV Cohort. Eur J Immunol 1997, 12:3223-3227.
66. Cohen OJ, Vaccarezza M, Lam GK, Baird BF, Wildt K, Murphy PM, et al. Heterozygosity for a defective gene for CC chemokine receptor 5 is not the sole determinant for the immunologic and virologic phenotype of HIV-infected long-term nonprogressors. J Clin Invest 1997, 100:1581-1589.
67. Migueles SA, Laborico AC, Imamichi H, Shupert WL, Royce C, McLaughlin M, et al. The differential ability of HLA B*5701+ long-term nonprogressors and progressors to restrict human immunodeficiency virus replication is not caused by loss of recognition of autologous viral gag sequences. J Virol 2003, 77:6889-6898.
68. Sheppard HW, Celum C, Michael NL, O'Brien S, Dean M, Carrington M, et al. HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J Acquir Immune Defic Syndr 2002, 29:307-313.
69. Guerin S, Meyer L, Theodorou I, Boufassa F, Magierowska M, Goujard C, et al. CCR5 delta32 deletion and response to highly active antiretroviral therapy in HIV-1-infected patients. AIDS 2000, 14:2788-2790. 70. Valdez H, Purvis SF, Lederman MM, Fillingame M, Zimmerman PA. Association of the CCR5delta32 mutation with improved response to antiretroviral therapy. JAMA 1999, 282:734.
71. O'Brien TR, McDermott DH, Ioannidis JP, Carrington M, Murphy PM, Havlir DV, Richman DD. Effect of chemokine receptor gene polymorphisms on the response to potent antiretroviral therapy. AIDS 2000, 14:821-826. 72. Wit FW, van Rij RP, Weverling GJ, Lange JM, Schuitemaker H. CC chemokine receptor 5 delta32 and CC chemokine receptor 2 64I polymorphisms do not influence the virologic and immunologic response to antiretroviral combination therapy in human immunodeficiency virus type 1-infected patients. J Infect Dis 2002, 186:1726-1732.
73. Brumme ZL, Chan KJ, Dong W, Hogg R, O'Shaughnessy MV, Montaner JS, Harrigan PR. CCR5Delta32 and promoter polymorphisms are not correlated with initial virological or immunological treatment response. AIDS 2001, 15:2259-2266.
74. Flores-Villanueva PO, Yunis EJ, Delgado JC, Vittinghoff E, Buchbinder S, Leung JY, et al. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci USA 2001, 98:5140-5145.
75. Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet 2002, 31:429-434.
76. Christiansen FT, Gaudieri S, Santis DD, Moore CB, Witt CS, James IR, Mallal SA. NK receptor genes are predictors of HIV progression. Hum Immunol 2003, 64 (Suppl):S12.
77. Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med 1996, 2:338-342.
78. Mallal S, Nolan D, Witt C, Masel G, Martin AM, Moore C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 2002, 359:727-732.
79. Hetherington S, Hughes AR, Mosteller M, Shortino D, Baker KL, Spreen W, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet 2002, 359: 1121-1122.
80. Martin AM, Nolan D, Gaudieri S, Almeida CA, Nolan R, James I, et al. Predisposition to Abacavir Hypersensitivity Conferred by HLA-B*5701 and a Haplotypic Hsp70-Hom variant. Proc Natl Acad Sci USA 2004; 101:4180-4185.
81. Patel SM, Johnson S, Belknap SM, Chan J, Sha BE, Bennett C. Serious adverse cutaneous and hepatic toxicities associated with nevirapine use by non-HIV-infected individuals. J Acquir Immune Defic Syndr 2004, 35:120-125.
82. Kontorinis N, Dieterich DT. Toxicity of non-nucleoside analogue reverse transcriptase inhibitors. Semin Liver Dis 2003, 23: 173-182.
83. Law WP, Dore GJ, Duncombe CJ, Mahanontharit A, Boyd MA, Ruxrungtham K, et al. Risk of severe hepatotoxicity associated with antiretroviral therapy in the HIV-NAT Cohort, Thailand, 1996-2001. AIDS 2003, 17:2191-2199.
84. Stern JO, Robinson PA, Love J, Lanes S, Imperiale MS, Mayers DL. A comprehensive hepatic safety analysis of nevirapine in different populations of HIV infected patients. J Acquir Immune Defic Syndr 2003, 34 (Suppl 1):S21-S33.
85. Schmitt M, Harrer E, Goldwich A, Bauerle M, Graedner I, Kalden JR, Harrer T. Specific recognition of lamivudine-resistant HIV-1 by cytotoxic T lymphocytes. AIDS 2000, 14:653-658. 86. Karlsson AC, Deeks SG, Barbour JD, Heiken BD, Younger SR, Hoh R, et al. Dual pressure from antiretroviral therapy and cell-mediated immune response on the human immunodeficiency virus type 1 protease gene. J Virol 2003, 77:6743-67452.
87. Oxenius A, Price DA, Dawson SJ, Gunthard HF, Fischer M, Perrin L, et al. Residual HIV-specific CD4 and CD8 T cell frequencies after prolonged antiretroviral therapy reflect pretreatment plasma virus load. AIDS 2002, 16:2317-2322. 88. Rinaldo CR Jr, Huang XL, Fan Z, Margolick JB, Borowski L, Hoji A, et al. Anti-human immunodeficiency virus type 1 (HIV-1) CD8(+) T-lymphocyte reactivity during combination antiretroviral therapy in HIV-1-infected patients with advanced immunodeficiency. J Virol 2000, 74:4127-4138.
89. Deeks SG, Martin JN, Sinclair E, Harris J, Neilands TB, Maecker HT, et al. Strong cell-mediated immune responses are associated with the maintenance of low-level viremia in antiretroviral-treated individuals with drug-resistant human immunodeficiency virus type 1. J Infect Dis 2004, 189:312-321.
90. Price DA, Scullard G, Oxenius A, Braganza R, Beddows SA, Kazmi S, et al. Discordant outcomes following failure of antiretroviral therapy are associated with substantial differences in human immunodeficiency virus-specific cellular immunity. J Virol 2003, 77:6041-6049.
91. Lacabaratz-Porret C, Urrutia A, Doisne JM, Goujard C, Deveau C, Dalod M, et al. Impact of antiretroviral therapy and changes in virus load on human immunodeficiency virus (HIV)-specific T cell responses in primary HIV infection. J Infect Dis 2003, 187:748-757.
92. Seth A, Markee J, Hoering A, Sevin A, Sabath DE, Schmitz JE, et al. Alterations in T cell phenotype and human immunodeficiency virus type 1-specific cytotoxicity after potent antiretroviral therapy. J Infect Dis 2001, 183:722-729.
93. Wood E, Hogg RS, Yip B, Quercia R, Harrigan PR, O'Shaughnessy MV, Montaner JS. Higher baseline levels of plasma human immunodeficiency virus type 1 RNA are associated with increased mortality after initiation of triple-drug antiretroviral therapy. J Infect Dis 2003, 188:1421-1425.
94. Chene G, Sterne JA, May M, Costagliola D, Ledergerber B, Phillips AN, et al. Prognostic importance of initial response in HIV-1 infected patients starting potent antiretroviral therapy: analysis of prospective studies. Lancet 2003, 362: 679-686.
95. Maggiolo F, Migliorino M, Pirali A, Pravettoni G, Caprioli S, Suter F. Duration of viral suppression in patients on stable therapy for HIV-1 infection is predicted by plasma HIV RNA level after 1 month of treatment. J Acquir Immune Defic Syndr 2000, 25: 36-43.
96. Paredes R, Mocroft A, Kirk O, Lazzarin A, Barton SE, van Lunzen J, et al. Predictors of virological success and ensuing failure in HIV-positive patients starting highly active antiretroviral therapy in Europe: results from the EuroSIDA study. Arch Intern Med 2000, 160:1123-1132.
97. Antinori A, Cingolani A, Giancola ML, Forbici F, De Luca A, Perno CF. Clinical implications of HIV-1 drug resistance in the neurological compartment. Scand J Infect Dis 2003, 35 (Suppl 106):41-44.
|
|
| |
| |
|
|
|