| |
Atazanavir enhances saquinavir hard-gel concentrations in a ritonavir-boosted once-daily regimen
|
| |
| |
AIDS: Volume 18(9) 18 June 2004 pp 1291-1297
Boffito, Martaa; Kurowski, Michaelb; Kruse, Guidob; Hill, Andrewc; Benzie, Andrew Aa; Nelson, Mark Ra; Moyle, Graeme Ja; Gazzard, Brian Ga; Pozniak, Anton La
From the aChelsea and Westminster Hospital, London, UK; bTherapia GmbH, Berlin, Germany; and cRoche, Welwyn, UK.
The authors say:
"...Although saquinavir/ritonavir/atazanavir (1600/100/300 mg once a day) was well tolerated by the highly treatment-experienced patients enrolled in this study, hyperbilirubinemia was quite common but reversible. Median serum concentrations of both total and unconjugated bilirubin increased by approximately fivefold by day 11 and by approximately sixfold by day 31. Hyperbilirubinemia was not felt to be clinically significant, and both total and unconjugated bilirubin returned to baseline by the follow-up visit. This observation was not unexpected as atazanavir is known to be a competitive inhibitor of uridine diphosphate-glucuronosyl transferase (ki 1.9 uM), the enzyme that conjugates bilirubin with glucuronide. Atazanavir is known to lead to unconjugated hyperbilirubinemia in a significant number of patients, and the likelihood of bilirubin elevations being over 2.5 mg/dl may also be proportional to the atazanavir AUC0-24. In the present study, six out of 18 patients (33%) were judged to have developed hyperbilirubinemia after the addition of atazanavir. Further studies of this regimen with lower doses of atazanavir may be warranted to examine the possibility of achieving therapeutic atazanavir levels without the development of hyperbilirubinemia- associated clinical manifestations... In conclusion, the data from this study indicate that saquinavir, ritonavir and atazanavir, dosed at 1600/100/300 mg once a day, exhibit favourable pharmacokinetic interactions and should therefore be considered for further clinical evaluation. The high saquinavir drug levels achieved by boosting with ritonavir were further increased with the addition of atazanavir. Atazanavir and ritonavir may use independent mechanisms to boost saquinavir..."
Abstract
Objective: To determine the pharmacokinetics of saquinavir hard-gel capsules/ ritonavir/atazanavir co-administered once daily at 1600/100/300 mg in HIV-infected individuals.
Methods: Eighteen patients receiving saquinavir/ritonavir switched to 1600/100 mg once daily a minimum of 3 days before the study. On study day 1, levels of saquinavir and ritonavir were determined over 24 h. Atazanavir (300 mg once daily) was then added to the regimen. On day 11, a pharmacokinetic analysis was performed. Atazanavir was discontinued on day 32. Drug concentrations were measured by high-pressure liquid chromatography-tandem mass spectrometry. Geometric mean ratios (GMR) and 95% confidence intervals (CI) were used to compare saquinavir and ritonavir pharmacokinetic parameters, with and without atazanavir. A safety analysis was performed at screening, days 1, 11, 32 and follow-up.
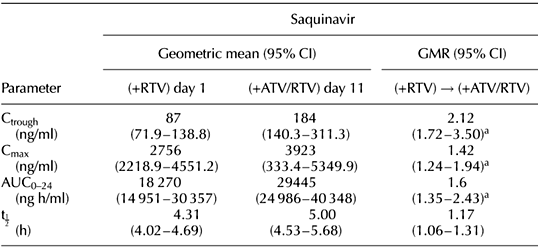
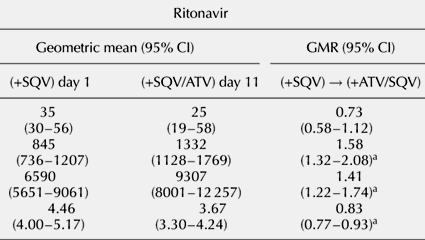
Results: After the addition of atazanavir, statistically significant increases in saquinavir trough plasma concentration (Ctrough GMR, 95% CI 2.12, 1.72-3.50), maximum plasma concentration (Cmax 1.42, 1.24-1.94), area under the plasma concentration-time curve from 0-24 h (AUC0-24 1.60, 1.35-2.43) and ritonavir Cmax (1.58, 1.32-2.08), AUC0-24 (1.41, 1.22-1.74) were observed.
The pharmacokinetics of atazanavir compared with those obtained in patients receiving atazanavir/ritonavir without saquinavir. Four patients developed scleral icterus and two jaundice. Total and unconjugated bilirubin increased approximately fivefold during atazanavir therapy.
Conclusion: The addition of atazanavir to saquinavir/ritonavir increased saquinavir Ctrough, Cmax and AUC0-24 by 112, 42 and 60%. Ritonavir Cmax and AUC 0-24 increased by 34 and 41%. The regimen was well tolerated, with no significant change in laboratory parameters, except for the occurrence of hyperbilirubinemia.
Safety and tolerability
The combination of saquinavir (1600 mg once a day) and ritonavir (100 mg once a day) co-administered with atazanavir (300 mg once a day) was generally well tolerated. The most common and most severe adverse event was hyperbilirubinemia (grade 2-4). Six patients (33%) developed hyperbilirubinemia after the addition of atazanavir: four (22%) developed scleral icterus and two (11%) developed jaundice. Fivefold increases were observed in the median total and unconjugated bilirubin levels between days 1 and 11. Total and unconjugated bilirubin levels remained elevated at day 31, but returned to baseline levels by follow-up.
All other adverse events were of mild (grade 1) severity only. During the saquinavir/ritonavir phase, one patient (5.5%) reported abdominal pain and one patient (5.5%) had a skin rash. After the addition of atazanavir, two patients (11%) each had diarrhoea and fatigue and one patient (5.5%) had headache.
Alanine transaminase, aspartate transaminase, total cholesterol, triglyceride and glucose levels all remained stable throughout the study.
Introduction
Protease inhibitor (PI) boosting has become an established strategy for enhancing the benefits of PI. This approach involves the concurrent administration of a potent cytochrome P450 (CYP) 3A4 inhibitor, usually a subtherapeutic dose of ritonavir, in order to enhance PI plasma concentrations. This may allow a reduction in the PI dose or dosing frequency, and may minimize the effects of food on bioavailability.
Saquinavir is frequently used as a boosted PI with low-dose ritonavir. A 1000/100 mg twice-daily dosage is approved in Europe and the USA, but clinical trials have indicated that once-daily saquinavir/ritonavir, at a dosage of 1600/100 mg, is also a viable therapeutic option [3,4]. Saquinavir/ritonavir (1600/100 mg once daily) plus two nucleoside reverse transcriptase inhibitors (NRTI) has recently shown comparable potency to efavirenz (600 mg once daily) plus two NRTI over 48 weeks in treatment-naive patients.
Atazanavir (formerly BMS-232632) is a new PI that is a substrate for CYP3A4, and co-administration with ritonavir significantly increases plasma atazanavir levels [6]. Atazanavir has good oral bioavailability and pharmacokinetic properties that allow once-daily dosing either with or without ritonavir [6]. However, unboosted atazanavir showed a significantly poorer viral load response than lopinavir/ritonavir at 24 weeks in a phase III study of atazanavir (AI424-043). Atazanavir, even when combined with ritonavir, has shown no apparent adverse effects on serum lipid levels
Saquinavir exposure is increased in the presence of atazanavir; however, a regimen of saquinavir/atazanavir 1200/400 mg is insufficient to achieve plasma trough levels consistently above the proposed minimum therapeutic trough concentration of saquinavir (100 ng/ml). These pharmacokinetic findings were reflected in the results of a phase III study in which a regimen of saquinavir/atazanavir 1200/400 mg once a day plus two NRTI was associated with poor treatment outcomes. The optimum dosage of saquinavir and atazanavir, when used in combination, has therefore yet to be established.
Atazanavir in vivo selects a unique I50L primary mutation in previously PI-naive individuals, with these isolates generally showing unchanged or enhanced susceptibility to saquinavir and other PI.
Given the positive pharmacokinetic interactions of atazanavir and saquinavir, their different primary resistance mutation patterns, their favourable lipid profiles and their suitability for once-daily dosing, a ritonavir-double-boosted saquinavir/atazanavir regimen may offer a potent, convenient treatment option with a good long-term safety profile. The aim of this study was therefore to evaluate the steady-state pharmacokinetics and safety over one month of a regimen of saquinavir/ritonavir/atazanavir 1600/100/300 mg once a day in HIV-1-infected adults.
Patients
After providing written informed consent 20 HIV-1-infected patients were enrolled in the study. These were adults (aged 19-65 years) of either sex who had been receiving a stable antiretroviral regimen containing saquinavir/ritonavir and two NRTI.
At screening (between day -28 and day -14) 16 patients had an HIV-RNA load of less than 50 copies/ml (branched DNA assay) and two patients had low levels of HIV RNA (57 and 61 copies/ml). All patients had a neutrophil count greater than 1000 cells/mm3, a platelet count of 75 000 or more, haemoglobin greater than 9.0 g/dl (men and women), alanine transaminase and aspartate transaminase less than 3.9 times the upper limit of normal (ULN), alkaline phosphatase less than 3.0 times the ULN, and total bilirubin less than 1.5 times the ULN.
None of the patients were permitted to take a non-NRTI, any treatment for an active infection (apart from HIV), any hepatic enzyme inducer or inhibitor within 14 days of study entry, or any investigational drug or antineoplastic radiotherapy/chemotherapy other than local skin radiotherapy within 12 weeks of starting study medication. The patients were allowed to receive other non-study medications provided that, in the opinion of the study investigators, these did not have any significant interaction with PI, and their doses had not changed by more than 25% in the preceding 28 days. Subjects were not allowed to commence new concomitant medications during the study period, which could alter the lipid profile or total and unconjugated bilirubin levels.
Study design
This was an open-label, single-arm study carried out at the Pharmacokinetics Unit of Chelsea and Westminster Hospital, London, UK. The study protocol was reviewed and approved by the Riverside Research Ethics Committee.
Patients entering the study had been receiving saquinavir/ritonavir for at least 2 weeks. A minimum of 3 days before the start of the study, patients were switched to saquinavir hard gel/ritonavir at a dosage of 1600/100 mg once a day (if they were not already receiving this formulation or dosage).
On day 1 all subjects received saquinavir/ritonavir 1600/100 mg once a day. From days 2 to 31 atazanavir 300 mg once a day was added to the regimen. Serial blood samples, for the determination of PI drug levels, were collected on days 1 and 11 pre-dose and 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h after the administration of the antiretroviral regimen with a standard 20 g fat meal.
The safety and tolerability of study medications were evaluated throughout the study on the basis of clinical adverse events (using the AIDS Clinical Trials Group toxicity grading scale to characterize abnormal findings), clinical laboratory tests (at screening, on days 1, 11 and 32, and at a follow-up visit), vital signs and physical examinations. The severity of adverse events and the investigator's assessment of their causality to saquinavir, ritonavir and atazanavir were recorded. The reference ranges for the clinical laboratory test used in this study were: total and unconjugated bilirubin 0-17 and 0-12 umol/l, alanine transaminase and aspartate transaminase 0-37 U/l, glucose 3.0-7.8 mmol/l, total cholesterol and triglyceride levels 3.5-6.5 and 0.00-2.20 mmol/l.
Results
Disposition of patients
Twenty HIV-1-positive patients were enrolled in the study, but two patients subsequently withdrew for non-drug-related reasons (one for psychiatric reasons and one because of urinary tract infection). The pharmacokinetic analysis is therefore based on the 18 subjects who completed the study. Their baseline characteristics were: two women, 16 men; mean age 41 years (range 22-57); and median CD4 cell count 442 cells/mm3 (118-947). Two patients (11.1%) had an HIV- RNA level greater than 50 copies/ml (57 and 61 copies/ml). The median body mass index was 23.3 kg/m2 (16.9-31.2).
The patients who completed the study received the following antiretroviral agents in addition to the study drugs: five were on zidovudine, 11 were on lamivudine, seven were on abacavir, five were on didanosine, one was on zalcitabine, one was on stavudine and six were on tenofovir disoproxil fumarate.
Pharmacokinetics TOP
Geometric mean values (95% CI) for the steady-state pharmacokinetic parameters for saquinavir, ritonavir and atazanavir are summarized in Table 1.
Table 1. Summary of geometric mean (95% confidence interval) steady-state pharmacokinetic parameters for saquinavir, ritonavir and atazanavir and mean change (geometric mean ratios and 95% confidence intervals) in saquinavir and ritonavir pharmacokinetic parameters between day 1 (saquinavir/ritonavir 1600/100 mg once a day) and day 11 (atazanavir/saquinavir/ritonavir 300/1600/100 mg once a daily).ATV, Atazanavir; AUC, area under concentration-time curve; CI, confidence interval; Cmax, maximum concentration; Ctrough, trough concentration; GMR, geometric mean ratio; RTV, ritonavir; SQV, saquinavir; t1⁄2, elimination half-life.aStatistically significant versus day 1.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
The saquinavir Ctrough, Cmax, AUC0-24 and t1⁄2 values observed on day 11 (in the presence of atazanavir) were increased by 112, 42, 60 and 17%, respectively, compared with those observed on day 1 (in the absence of atazanavir). These differences were statistically significant (Fig. 1).
The ritonavir Cmax and AUC0-24 values observed on day 11 (in the presence of atazanavir) were also significantly increased by atazanavir co-administration, by 34 and 41%, respectively. The ritonavir Ctrough and t1⁄2 values observed on day 11, however, were decreased by 27 and 25%, respectively, compared with those observed on day 1. The decrease in t1⁄2 was statistically significant, but the decrease in Ctrough was not (Fig. 2).
Discussion
This open-label, single-arm study was designed to determine the steady-state pharmacokinetics of once-daily saquinavir (1600 mg) and ritonavir (100 mg) with and without atazanavir (300 mg) when administered to HIV-1-positive adults.
The addition of atazanavir to a regimen containing saquinavir/ritonavir substantially increased saquinavir exposure, producing increases in the saquinavir Ctrough, Cmax, AUC0-24 and t1⁄2 that were statistically significant. Moreover, the addition of atazanavir also influenced the pharmacokinetics of ritonavir. The ritonavir Cmax and AUC0-24 were significantly increased, but the ritonavir Ctrough and t1⁄2 were slightly decreased.
The combination of saquinavir with atazanavir did not appear to influence exposure to atazanavir because the pharmacokinetics of atazanavir observed in the present study are in accordance with previously published values [16]. The geometric mean Ctrough, Cmax and AUC0-24 values of atazanavir observed in the present study (767 ng/ml, 4982 ng/ml and 51 036 ng h/ml, respectively) were also similar to the geometric mean Cmin, Cmax and AUC0-24 values of atazanavir observed when atazanavir/ritonavir was administered to HIV-1-infected patients at a dose of 300/100 mg once a day (636 ng/ml, 4422 ng/ml and 46 073 ng h/ml).
Reductions in atazanavir Cmin and AUC0-24 have been reported when tenofovir is co-administered with atazanavir (both with and without ritonavir boosting). In this study, one-third of subjects were on concomitant tenofovir. In the six subjects receiving tenofovir, the median atazanavir Ctrough level was observed to be slightly higher than in the 12 subjects who were not (974 versus 757 ng/ml), whereas the median AUC0-24 was similar (48 684 versus 55 196 ng/ml). However, this study was not designed or powered to measure the impact of tenofovir on atazanavir levels.
The mechanism by which atazanavir boosts saquinavir and ritonavir is unknown. Increased ritonavir levels could have resulted in the greater inhibition of CYP3A4, with a consequent increase in saquinavir levels. However, it has previously been shown that increasing the concentration of ritonavir given with saquinavir above 100 mg does not further increase saquinavir exposure. Furthermore, this explanation would not account for the mechanism by which atazanavir increased ritonavir levels. It is unlikely that the inhibition of CYP3A4 by atazanavir resulted in the significant increase in ritonavir and saquinavir levels, as ritonavir is a more potent competitive inhibitor of CYP3A4 in human liver microsomes (Ki 0.019 uM) than ATZ (2.35 uM). Isoforms other than CYP3A4 are also unlikely to be the mechanism as it has not yet been possible to demonstrate the inhibition of saquinavir by isoforms other than CYP3A4. PI have been shown to be substrates and inhibitors of P-glycoprotein.
We, therefore, speculate that the inhibition of P-glycoprotein-mediated drug transport may be responsible for the increase in saquinavir and ritonavir exposure, such that when atazanavir, saquinavir and ritonavir are co-administered, ritonavir predominantly inhibits CYP3A4, increasing the exposure of both atazanavir and saquinavir, whereas atazanavir predominantly inhibits P-glycoprotein-mediated drug transport, increasing both ritonavir and saquinavir levels. We have found no data in the literature on the effect of atazanavir on P-glycoprotein drug transport, and so this suggested explanation for the observed results is, at this stage, purely hypothetical. However, potent P-glycoprotein inhibitors have been shown to increase PI concentrations in both HIV sanctuary sites and plasma in mice.
Although saquinavir/ritonavir/atazanavir (1600/100/300 mg once a day) was well tolerated by the highly treatment-experienced patients enrolled in this study, hyperbilirubinemia was quite common but reversible. Median serum concentrations of both total and unconjugated bilirubin increased by approximately fivefold by day 11 and by approximately sixfold by day 31. Hyperbilirubinemia was not felt to be clinically significant, and both total and unconjugated bilirubin returned to baseline by the follow-up visit. This observation was not unexpected as atazanavir is known to be a competitive inhibitor of uridine diphosphate-glucuronosyl transferase (Ki 1.9 uM), the enzyme that conjugates bilirubin with glucuronide. Atazanavir is known to lead to unconjugated hyperbilirubinemia in a significant number of patients, and the likelihood of bilirubin elevations being over 2.5 mg/dl may also be proportional to the atazanavir AUC0-24. In the present study, six out of 18 patients (33%) were judged to have developed hyperbilirubinemia after the addition of atazanavir. Further studies of this regimen with lower doses of atazanavir may be warranted to examine the possibility of achieving therapeutic atazanavir levels without the development of hyperbilirubinemia- associated clinical manifestations.
The elevations of serum bilirubin levels sometimes associated with atazanavir therapy are thought not to be a manifestation of drug-induced hepatic injury. Consistent with this, there were no discernable changes in the median levels of either alanine transaminase or aspartate transaminase throughout the present study. Although two patients (11%) in the present study experienced jaundice and four (22%) developed scleral icterus, the study population was too small to assess reliably whether these adverse events were more common than previously observed.
In this study, after the addition of atazanavir to an existing saquinavir/ritonavir regimen, there were no discernable changes in the median levels of total cholesterol, triglycerides or glucose.
In conclusion, the data from this study indicate that saquinavir, ritonavir and atazanavir, dosed at 1600/100/300 mg once a day, exhibit favourable pharmacokinetic interactions and should therefore be considered for further clinical evaluation. The high saquinavir drug levels achieved by boosting with ritonavir were further increased with the addition of atazanavir. Atazanavir and ritonavir may use independent mechanisms to boost saquinavir.
|
|
| |
| |
|
|
|