| |
Steady-State Pharmacokinetics of Saquinavir Hard-Gel/Ritonavir/Fosamprenavir in HIV-1-Infected Patients
|
| |
| |
JAIDS Journal of Acquired Immune Deficiency Syndromes: Volume 37(3) 1 November 2004
Boffito, Marta MD, PHD*; Dickinson, Laura BSC†; Hill, Andrew PHD‡; Back, David PHD†; Moyle, Graeme MD*; Nelson, Mark MD*; Higgs, Chris*; Fletcher, Carl*; Gazzard, Brian MD*; Pozniak, Anton MD*
From the *Chelsea and Westminster Hospital, London; †University of Liverpool; and ‡Roche, Welwyn, UK.
Abstract
Background: In vitro synergy and complementary resistance profiles provide a strong rationale for combining fosamprenavir with saquinavir as part of a potent double-boosted protease inhibitor regimen. This study evaluated the steady-state pharmacokinetics of saquinavir 1000 mg twice daily (bid) and fosamprenavir 700 mg bid administered with 2 different doses of ritonavir (100 and 200 mg bid) in HIV-1-infected subjects.
Methods: On day 1, 12-hour pharmacokinetic profiles for saquinavir/ritonavir (1000/100 mg bid) were obtained for 18 subjects. All subjects were receiving ongoing treatment with a saquinavir/ritonavir-containing regimen. Fosamprenavir 700 mg bid was then added to the regimen, and pharmacokinetic sampling was repeated for all 3 agents at day 11. The ritonavir daily dose was then increased to 200 mg bid, and a 3rd pharmacokinetic profile was obtained at day 22.
Results: The coadministration of fosamprenavir 700 mg bid with saquinavir/ritonavir 1000/100 mg bid resulted in a statistically nonsignificant decrease in saquinavir concentrations (by 14, 9, and 24%, for saquinavir area under the concentration-time curve [AUC]0-12, Cmax, and Ctrough, respectively). This was compensated for by an increased ritonavir dose of 200 mg bid, which resulted in a statistically nonsignificant increase in saquinavir exposure compared with baseline. Amprenavir levels did not appear to be significantly influenced by coadministration of saquinavir with fosamprenavir. Fosamprenavir significantly reduced ritonavir exposure, but the increased ritonavir dose compensated for this interaction.
Conclusions: Our findings showed that saquinavir/ritonavir/fosamprenavir was well tolerated over the study period. Saquinavir plasma concentrations were slightly lowered by the addition of fosamprenavir to the regimen. However, the addition of a further 100 mg ritonavir bid restored the small and insignificant decrease.
BACKGROUND
A range of different strategies aimed at managing HIV-infected patients who have experienced treatment failure has been explored in clinical practice, including the administration of ritonavir-boosted protease inhibitor (PI) combinations. The inclusion of a single-boosted PI as part of an antiretroviral regimen in moderately PI-experienced patients has met with only limited success in clinical trials, with an undetectable viral load (HIV RNA levels <50 copies/mL) achieved in approximately half of patients at 24 weeks.
A combination of 2 active PIs may extend the spectrum of activity against HIV and increase regimen potency, and there is evidence from clinical trials that dual-PI-containing regimens may lead to greater antiviral efficacy and a more favorable treatment outcome in PI-experienced patients than a single-PI-containing regimen. However, all the currently approved PIs are metabolized by cytochrome P450 (predominantly CYP3A4), and thus clinically relevant and sometimes complex drug-drug interactions can occur when these agents are coadministered. For example, coadministration of the fixed-dose combination of lopinavir/ritonavir (Kaletra; Abbott Laboratories, Abbott Park, IL) with amprenavir results in significantly lower plasma concentrations of both lopinavir and amprenavir in HIV-infected individuals than those observed in historical controls.
The HIV-1 PI fosamprenavir-a prodrug of amprenavir-is rapidly hydrolyzed to its parent form by cellular phosphatases in the gut epithelium during absorption. The safety and efficacy of fosamprenavir have been demonstrated over 48 weeks in both antiretroviral-naive and treatment-experienced patients. Moreover, fosamprenavir appears to be associated with fewer clinical adverse events than amprenavir, and the pill count associated with fosamprenavir is significantly lower than with amprenavir (4 vs. 16 capsules/d). Amprenavir undergoes CYP450-based metabolism in the gastrointestinal tract and liver and has been shown to both inhibit and induce different isoforms of this system. Coadministration of ritonavir, a potent CYP450 inhibitor, decreases the elimination of amprenavir, resulting in clinically significant increases in amprenavir exposure; thus, reduced doses of both agents are recommended when they are prescribed in combination. Amprenavir plasma levels are boosted in a similar way when ritonavir is coadministered with fosamprenavir. However, despite the presence of low doses of ritonavir, the coadministration of fosamprenavir and lopinavir/ritonavir leads to a reduction of both amprenavir and lopinavir plasma exposure.
Saquinavir, a widely used HIV-1 PI, is also a substrate of CYP450. Studies have shown that coadministration with low-dose ritonavir increases the area under the plasma concentration-time curve and peak concentration of saquinavir by >50-fold and 20-fold, respectively. Saquinavir boosted by ritonavir 1000/100 mg twice daily (bid) has shown clinical efficacy in reverse transcriptase inhibitor (RTI)-containing regimens in both PI-naive and moderately PI-experienced patients over 48 weeks. This dosage is now approved in both the United States and Europe.
Because amprenavir exhibits synergistic anti-HIV-1 activity with saquinavir in vitro and the 2 agents have nonoverlapping primary resistance patterns, there is a strong rationale for using fosamprenavir with saquinavir as part of a potent dual-PI regimen. Although the impact of fosamprenavir administration on the pharmacokinetics of saquinavir has not been fully elucidated, amprenavir coadministration was found to nonsignificantly reduce saquinavir exposure by approximately 20% compared with historical data for saquinavir alone from one study of HIV-infected subjects. It is conceivable that the pharmacokinetic enhancement of saquinavir by ritonavir may be able to compensate for the effects of CYP450 induction by amprenavir. However, the addition of amprenavir to saquinavir/ritonavir has been shown to reduce both ritonavir and saquinavir exposure significantly. The optimal ritonavir dose to compensate for the effects of amprenavir on saquinavir exposure has yet to be determined.
The aim of this study was therefore to investigate the steady-state pharmacokinetics of a regimen containing both saquinavir 1000 mg bid and fosamprenavir 700 mg bid administered with 2 different doses of ritonavir-100 mg bid and 200 mg bid-in HIV-1-infected individuals.
Subjects
Subjects eligible for this study were HIV-1 antibody-seropositive adults who were 19-65 years of age, were receiving ongoing treatment with a saquinavir/ritonavir-containing regimen, and had an undetectable plasma viral load (<50 HIV-1 RNA copies/mL; AMPLICOR HIV-1 MONITOR Test, Roche Diagnostics, Basel, Switzerland). Exclusion criteria included any active, clinically significant disease or any grade 3 or 4 toxicity according to the AIDS Clinical Trial Group (ACTG) grading severity list, other than grade 3 or 4 asymptomatic triglyceride/cholesterol elevations. Subjects were also ineligible for enrollment if they had taken either corticosteroids systemically or drugs known to induce or inhibit hepatic enzymes within 14 days of study entry. Approval for the study was obtained from the local ethics committee (Riverside Research Ethics Committee) and all subjects gave written informed consent to participate in the study.
Study Design and Procedures
This open-label, single-arm, pharmacokinetic study was performed at a single UK site (PK Research, Ltd., Chelsea and Westminster Hospital, London). A minimum of 3 days before the baseline visit (day 1), all subjects on other formulations of saquinavir were switched to saquinavir 200 mg hard-gel capsules (1000 mg bid coadministered with ritonavir at a dose of 100 mg bid), which was the formulation used throughout the study period. Study medications were administered as part of a combination antiretroviral regimen containing 2 nucleoside/nucleotide RTIs.
On day 1, subjects attended the study unit for pharmacokinetic sampling. On day 2, subjects added fosamprenavir 700 mg bid to their daily treatment regimen and on day 11 they returned to the unit for pharmacokinetic sampling. On day 12, the study regimen was again revised, this time to include an increased ritonavir dose of 200 mg bid. The final pharmacokinetic sampling day was day 22.
On the 3 pharmacokinetic assessment days (day 1, day 11, and day 22), study participants received a standardized breakfast containing 20 g of fat just before ingestion of their morning dose of study medications. Ingestion of all drug doses was directly observed and timed by study staff to ensure an accurate assessment of the interval between drug intake and blood sampling. Venous blood samples were collected from study subjects before dose administration and 0.5, 1, 2, 3, 4, 6, 8, 10, and 12 hours following ingestion. Blood samples were collected into heparinized tubes, centrifuged, and then stored at -70°C until analysis.
Safety Assessment
The safety and tolerability of the study medications were evaluated at each study visit on the basis of clinical adverse events (using the ACTG toxicity grading scale to characterize abnormal findings), clinical laboratory tests, vital signs, and physical examinations. The severity of adverse events and the investigator's assessment of their causality to study drugs were recorded.
RESULTS
Eighteen HIV-1-infected subjects (1 female) participated in the study and completed all phases of the study. Median (range) age and body mass index were 43 (22-57) years and 22 (17-29), respectively. At screening, median CD4+ cell count was 385 (153-806) and plasma HIV RNA was undetectable in 16/18 subjects (when detectable, n = 2, the viral load was 85 and 117 copies/mL). Concurrent antiretroviral medications administered with the study medications included zidovudine (n = 4), lamivudine (n = 6), stavudine (n = 1), didanosine (n = 6), abacavir (n = 7), zalcitabine (n = 1), and tenofovir diproxil fumarate DF) (n = 8).
Both the saquinavir/ritonavir/fosamprenavir regimens that were evaluated were well tolerated by study subjects, with no grade 3 or 4 adverse events reported. Study subjects were already established on saquinavir/ritonavir regimens; adverse events that were potentially drug related and that occurred when fosamprenavir was added to the regimen were nausea (mild, n = 1; moderate, n = 1), fatigue (mild, n = 2; moderate, n = 1), and diarrhea (mild, n = 1). Liver function tests and total cholesterol concentrations remained stable over the study period, with the exception of 1 subject who experienced a relevant increase in aspartate aminotransferase (379 U/L) and alanine aminotransferase (780 U/L) as a result of hepatitis C reactivation (hepatitis C virus polymerase chain reaction = 40,000 copies/mL) during the study period.
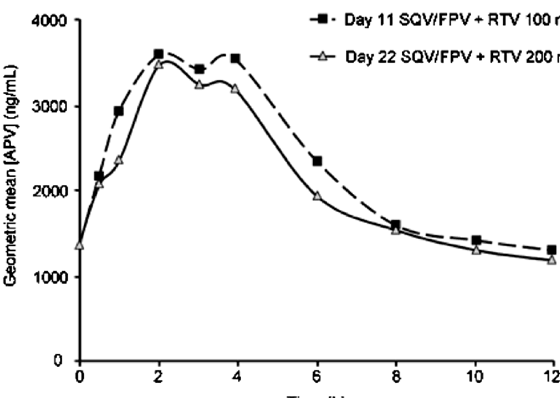
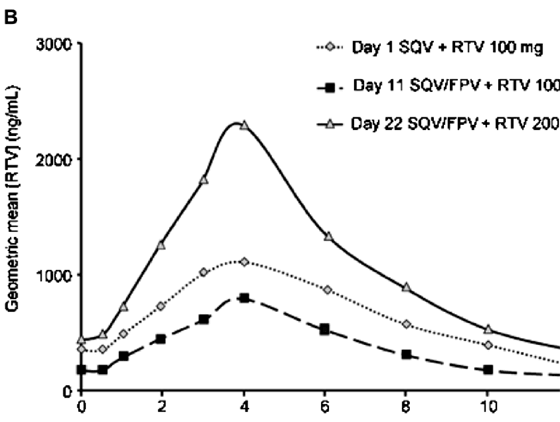
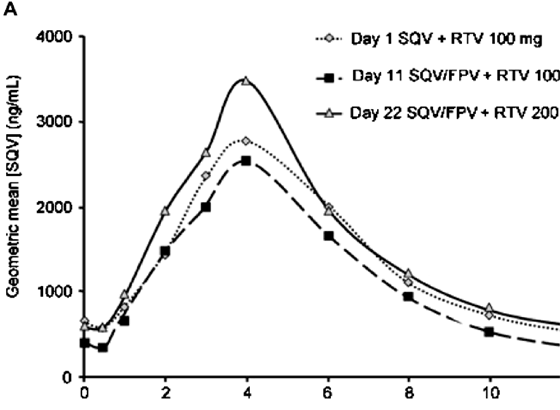
Figure 1 shows the plasma concentration-time curves for saquinavir, ritonavir, and amprenavir over the 12-hour dosing period during steady-state conditions for the 3 evaluated dosing regimens. On day 11 (ritonavir 100 mg bid), the saquinavir AUC (GMR, 95% CI: 0.85, 0.74-1.15), Cmax (0.91, 0.79-1.18), and Ctrough (0.76, 0.64-1.17) showed a statistically nonsignificant decrease from baseline. On day 22 (ritonavir 200 mg bid), the saquinavir AUC (GMR, 95% CI: 1.09, 0.87-1.65), Cmax (1.19, 0.97-1.67), and Ctrough (1.02, 0.81-1.74) showed a statistically nonsignificant increase compared with baseline.
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
Amprenavir AUC, Cmax, and Cmin day 11 values were 27, 29, and 40% lower than those seen for historical controls. At days 11 and 22, 17/18 patients had an amprenavir Ctrough that was higher than the recommended minimum trough levels for wild-type HIV (400 ng/mL) (geometric mean 1299 ng/mL, day 11, and 1152 ng/mL, day 22). The additional ritonavir boost of 200 mg/d in the 2nd study phase had no significant effect on plasma levels of amprenavir, with comparable values for each of the pharmacokinetic parameters investigated (GMR, 95% CI, for day 22 vs. day 11: AUC 0.92, 0.85-1.01; Cmax 0.97, 0.87-1.16; Ctrough 0.89, 0.69-1.29).
On day 11, ritonavir pharmacokinetic parameters were significantly lower after the addition of fosamprenavir to the regimen (GMR, 95% CI: AUC 0.60, 0.53-0.73; Cmax 0.71, 0.63-0.86; Ctrough 0.46). However, the subsequent increase in ritonavir dose from 100 mg bid to 200 mg bid more than compensated for this interaction, resulting in significantly higher ritonavir plasma concentrations at day 22 than at baseline (GMR, 95% CI: AUC 1.72, 1.50-2.15; Cmax 2.10, 1.83-2.67; Ctrough 1.37, 1.16-1.90).
Although subjects received PI doses after a standardized meal on pharmacokinetic study days, the interpatient variability among AUC, Cmax, and Ctrough values was high. The CVs for saquinavir Ctrough values were 90, 75, and 102% for saquinavir/ritonavir 1000/100-mg, saquinavir/ritonavir/fosamprenavir 1000/100/700-mg, and saquinavir/ritonavir/fosamprenavir 1000/200/700-mg regimens, respectively. However, saquinavir Ctrough values (geometric mean) were above the recommended minimum trough level of 100 ng/mL39 for all 3 regimens, and only 1 of the 18 subjects had saquinavir Ctrough values <100 ng/mL on days 11 and 22 (as well as on day 1).
AUTHOR DISCUSSION
We report here the safety and steady-state pharmacokinetics of 2 twice-daily, double-boosted PI regimens containing saquinavir and the amprenavir prodrug fosamprenavir (1000/700 mg bid) boosted by ritonavir at a dose of either 100 mg bid or 200 mg bid. The coadministration of fosamprenavir with saquinavir/ritonavir 1000/100 mg bid resulted in a modest decrease in saquinavir concentrations, but this was overcome by increasing the ritonavir dose to 200 mg bid. Amprenavir levels did not appear to be significantly influenced by coadministration of saquinavir with fosamprenavir. Both regimens were well tolerated, with adverse events limited to a small number of study subjects who reported grade 1 or 2 nausea, fatigue, or diarrhea after the addition of fosamprenavir to the saquinavir/ritonavir regimen.
A number of double-boosted PI regimens, which use ritonavir to boost levels of a dual-PI combination, have also been explored. Amprenavir/lopinavir/ritonavir regimens are complicated by the negative drug interaction between amprenavir and lopinavir/ritonavir, which could lead to a decreased virologic response, but promising results have been seen in heavily pretreated patients with double-boosted saquinavir regimens, such as saquinavir/lopinavir/ritonavir, both with and without additional RTIs.
Two small pilot trials of the double-boosted saquinavir/ritonavir/amprenavir combination in treatment-experienced, HIV-infected subjects have been reported. In one study (n = 10) the authors concluded that amprenavir and saquinavir did not appear to adversely influence plasma levels of each other when coadministered with ritonavir, while in the second study (n = 5), both saquinavir levels and ritonavir levels appeared to be significantly decreased following the addition of amprenavir to the regimen; amprenavir pharmacokinetics were not significantly influenced by the addition of saquinavir. Despite this interaction, a favorable response in HIV RNA and CD4 cell count was observed at 4 weeks, supporting further investigation of optimal dosing strategies for coadministration of fosamprenavir with saquinavir/ritonavir.
In our study, the coadministration of fosamprenavir 700 mg bid with saquinavir/ritonavir 1000/100 mg bid resulted in a modest decrease in saquinavir concentrations (by 14, 9, and 24%, for saquinavir AUC, Cmax, and Ctrough, respectively), but these were compensated for by an increase in ritonavir dose to 200 mg bid, which increased saquinavir pharmacokinetic values compared with baseline by 12, 20, and 3% (for AUC, Cmax, and Ctrough, respectively).
Compared with control data from a previous trial of fosamprenavir/ritonavir,8 amprenavir AUC, Cmax, and Cmin values were decreased by 27, 29, and 40%, respectively, in this study. However, 17/18 patients maintained Ctrough levels above the therapeutic threshold recommended for treatment-naive patients. It is not possible to determine from this study whether the lower amprenavir levels were simply due to interstudy variation or whether they are a result of the coadministration of saquinavir. Interestingly, a decrease in amprenavir exposure also occurred when amprenavir was coadministered with saquinavir soft-gel capsule32; however, whether this is clinically relevant is still unclear.
The effect of amprenavir on ritonavir plasma concentrations has not been clearly enumerated from past studies. In this study, a 54% decrease from baseline in ritonavir Ctrough was observed with the addition of fosamprenavir. It is unclear whether this reduction may have contributed to the reduction in saquinavir levels seen when fosamprenavir was added to the regimen.
Reductions in some PI (ie, atazanavir) Ctrough and AUC0-24 values have been reported when tenofovir DF is coadministered (both with and without ritonavir boosting). In this study, 8 subjects were on concomitant tenofovir DF. In these 8 subjects, the median saquinavir and fosamprenavir Ctrough and AUC0-12 were observed to be slightly higher than in the 10 subjects who were not treated with tenofovir DF (985, 457, and 729 ng/mL vs. 576, 396, and 458 ng/mL; and 29,038; 16,367; and 26,523 ng · h/mL vs. 19,813; 17,269; and 17,875 ng · h/mL for saquinavir on days 1, 11, and 22, respectively; and 1839 and 1749 ng/mL vs. 962 and 1096 ng/mL; 31,121; and 31,871 ng · h/mL vs. 26,833 and 24,024 ng · h/mL for amprenavir on days 11 and 22). However, this study was not designed or powered to measure the impact of tenofovir DF on PI concentrations.
Adherence to antiretroviral therapy is fundamental to achieve and maintain virologic response. Drug manufacturers are responding by developing new agents and formulations of existing drugs that simplify HIV treatment. Fosamprenavir, which has a significantly lower pill count than amprenavir, has been approved by the Food and Drug Administration for both once-daily (not recommended for PI-experienced patients) and twice-daily dosing when coadministered with ritonavir, and a number of once-daily boosted saquinavir regimens are under evaluation, with promising results to date. Moreover, a new 500-mg capsule formulation of saquinavir is in development and will reduce the saquinavir pill count from 5 to 2 capsules per 1000-mg dose.
In summary, our findings showed that the double-boosted PI combination saquinavir/ritonavir/fosamprenavir was well tolerated over the study period. Saquinavir plasma concentrations were slightly lowered by the addition of fosamprenavir to the regimen. However, the addition of a further 100 mg of ritonavir bid restored the small and insignificant decrease.
The study provides reassuring pharmacokinetic data particularly for patients who do not have RTIs as a treatment option, either because RTI-based regimens have failed them or they are intolerant to such regimens. Future long-term studies to further evaluate the safety and investigate the efficacy of this combination are warranted.
|
|
| |
| |
|
|
|