 |
|
|
| |
Gains in Understanding Drug-Resistant HIV
|
| |
| |
--Transmission of Drug Resistance Up; Viral Blips; Avoid the K65R Mutation; drug resistance increases if you stay on regimen with detectable viral load--
Report 5 from the 44th ICAAC, October 30-November 2, 2004, Washington, DC
Written for NATAP by Mark Mascolini
The ICAAC meeting had only a handful of reports on resistance to antiretrovirals, but they were enlightening. Transmission of drug-resistant HIV, which seemed to stabilize in the United States over recent years, took a big leap in a survey of 40 US cities. A small study failed to link 'blips' in viremia to resistance, but another inquest confirmed that incompletely suppressive regimens breed resistant virus. Two studies shed more light on emergence of the tenofovir-induced K65R mutation.
Transmission of drug-resistant HIV
Several years ago a ground-breaking study of 377 treatment-naive people in 10 North American cities pegged the rate of high-level resistance to one or more drugs at 12.4% in 1999-2000 (1). Later work by those same researchers suggested that transmission of resistant virus stabilized around that level (2). A 1997-2001 survey of 1082 untreated people in 10 US cities put the rate of transmitted reverse transcriptase and major protease mutations at 8.3% (3). But the newest study of untreated Americans with HIV infection reckoned that nearly one quarter have reduced susceptibility to one or more antiretrovirals (4). This survey by GlaxoSmithKline's Lisa Ross involved 317 untreated people seen for HIV infection at 54 clinics in 40 US cities. Researchers gathered all samples in 2003 and rated them as susceptible or resistant to specific drugs according to ViroLogic's April 2004 cutoffs. Most study participants (92%) were men, 55% were white, 33% were black, and 12% Hispanic. About three quarters picked up HIV during gay sex, while the rest got infected heterosexually of by injecting drugs. A little more than one third had fewer than 200 CD4 cells/µL. Seventy-three of the 317 people studied (23%) had virus with reduced susceptibility to one or more antiretrovirals. As in earlier studies, resistance to nonnucleosides proved more frequent than resistance to nucleosides or protease inhibitors:
|
|
| |
| |
 |
|
| |
| |
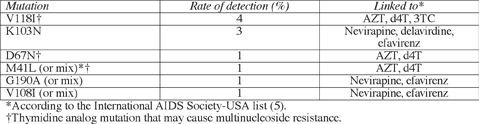
When Ross and colleagues looked only at primary resistance mutations, they counted them in 54 people (14%)—still a higher rate than in other US studies (1-3). The most common individual mutations were the mult-inucleoside resistance mutation V118I and the non-nucleoside killer K103N:
|
|
| |
| |
 |
|
| |
| |
No PI mutation appeared in 1% or more of the samples studied. Virus with reduced susceptibility to antiretrovirals proved more common among whites (41 of 175, or 27%) and blacks (24 of 106, or 23%) than among Hispanics (2 of 36, or 6%). But prevalence of one or more resistance mutations did not differ by race or ethnicity. Resistant virus showed up in 52 of 218 men (24%) infected during gay sex and in 14 of 75 people (19%) infected during sex between men and women.
As the HIV epidemic continues to evolve—in the US and anywhere antiretrovirals get used—emergence of resistant virus in treatment-naive people will continue to evolve apace.
Do blips mean trouble or not?
The meaning of blips—intermittently measurable virus in people with otherwise undetectable loads—continues to dog HIV clinicians. Are blips inconsequential jumps in HIV load, maybe spillover from a viral sanctuary? Or are they signals that ongoing viral replication below the 50-copy limit is getting out of hand? A new study presented by Richard Nettles from Robert Siliciano's group at Johns Hopkins suggests another possibility: Blips may just reflect variability in the assays that spot them (6). Three earlier studies found that blips to do not portend a permanent jump in viral load and virologic failure (7-9). But one study saw a higher risk of sustained failure after a single blip (10), and another saw consecutive blips as a harbinger of failure (11). Nettles linked blips neither to failure nor to resistance. To get a better handle on how often blips actually surface, Nettles measured viral loads three times a week for 3 to 4 months with the Roche version 1.5 assay in 10 people who had kept their viral load under 50 copies/mL for 11 to 79 months. Five were taking a PI regimen, 3 were using a nonnucleoside, and 2 were taking both a PI and a nonnucleoside. Two independent labs read the viral load results. Before the study's intensive monitoring phase, these 10 people had six blips in 125 viral load assays (4.8%). The 3-times-weekly monitoring recorded a slightly lower blip rate—26 in 713 assays, or 3.6%. One person had no blips during the study period. The monthly blip rate averaged 0.6 and ranged from 0 to 1.67, while the average blip size measured 87.4 copies/mL with a range from 51 to 201 copies/mL. The 3-times-weekly monitoring let Nettles calculate a maximum average blip duration of 92 hours, ranging from 48 to 276 hours. Ultrasensitive genotyping uncovered only one new mutation after a blip, the M46I mutation induced by protease inhibitors. That mutation did not appear in later assays. Because the person in whom M46I appeared had once taken an unsuccessful PI regimen, Nettles and colleagues suspect the mutation evolved earlier and continued to circulate at undetectable levels. The number of blips each person had—none, one, or two—didn't correlate with any of the variables that Nettles considered:
• Gender
• Race
• Age
• HIV risk factor
• CD4-cell nadir
• Duration of HIV infection
• Duration of virologic suppression
• Type of antiretroviral regimen
• Intercurrent illness or influenza vaccination
The Johns Hopkins team concluded that most people with a sub-50-copy viral load have blips, but that blips rarely last long enough to allow resistant virus to evolve. Notably, the independent labs that read viral load assays disagreed regularly on whether individual readings were above or below 50 copies/mL. That high discordance rate led Nettles and colleagues to propose that most blips represent normal viral load assay variation rather than clinically significant jumps in HIV RNA.
Resistance rate with a "stable" regimen
Whereas viral blips may not open the door to resistant virus (see preceding item), ongoing low-level viremia does. The question is how fast resistant virus pops up, and how quickly to switch antiretrovirals in an attempt to head off resistance. Some clinicians remain reluctant to change regimens quickly, fearing they may "use up" too many antiretrovirals too quickly with speedy antiretroviral swaps. Besides, they observe, CD4 counts often remain stable or even keep climbing after viral loads edge up into detectable territory. Some research shows, however, that sticking with an incompletely suppressive regimen gives resistant virus a green light (12,13). And antiretroviral options actually get "used up" faster when resistance appears. A new analysis of 98 people with low-level viral loads while taking a "stable" regimen confirmed this heightened risk of resistance (14). D. Edwards and coworkers at the University of North Carolina tracked viral evolution in 98 people who had two genotypic assays for resistant virus done more than 30 days apart. The median time between the two genotype assays was 9.3 months. All had taken the same antiretroviral for at least 30 days before the first genotype and continued the same combination through the second genotype.
The 98 study participants had taken antiretrovirals for a median 4 years and had taken their current regimen for a median 8.9 months before the first genotype in this analysis. Most (55%) were taking a PI-based regimen, while 14% were taking a nonnucleoside, 14% a PI and a nonnuke, and 16% nucleosides only. The group's median viral load bounced around the 10,000-copy mark from the time they started their current regimen through the first and second genotypes:
• Viral load at start of regimen: 4.2 logs (about 15,850 copies/mL), interquartile range (IQR) 3.0 to 5.0 logs
• Viral load at first genotype: 3.9 logs (about 7950 copies/mL), IQR 3.1 to 4.6 logs
• Viral load at second genotype: 4.3 logs (about 19,950 copies/mL), IQR 3.5 to 4.9 logs
When these 98 people had their first genotype, 86 (88%) had at least one resistance mutation and the median number was three. When they had their second genotype, 91 (93%) had at least one mutation and the median number was four. New mutations arose in 59 people (60%): 27% picked up one new mutation, 12% picked up two, and 21% picked up three.
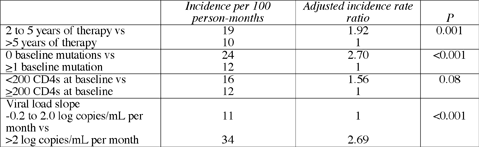
Among people with no evidence of nucleoside resistance on the first genotype, 40% had nucleoside mutations on the second genotype. Among those with no nonnucleoside resistance on the first test, 67% had it on the second. And among people with no PI resistance on the first genotype, 43% had resistance to a PI on their second genotype. Edwards calculated that these people gathered 13 new mutations per 100 person-months of follow-up or about 1.5 new mutations per year. Gender, race, or age did not influence the rate at which new mutations emerged. Neither did the current type of antiretroviral therapy, months of current therapy before the first genotype, or the viral load at the first genotype. But a rising viral load during follow-up and three other factors did predict the emergence of new mutations:
|
|
| |
| |
 |
|
| |
| |
Who runs the biggest risk of getting K65R?
The K65R mutation has captivated the interest of HIV virologists and clinicians for three reasons: First, since tenofovir disoproxil fumarate (TDF) came into wide use, the prevalence of this formerly rare mutation has been on the rise (15,16). Second, the mutation emerged regularly in studies of failing TDF regimens that also included abacavir and 3TC (17-19) or ddI and 3TC (20). Third, when K65R does appear, it can thwart the response not only to TDF, but also to ddI, d4T, 3TC, abacavir, and FTC (5). So figuring out which people taking TDF run the biggest risk of getting K65R could help improve antiretroviral planning. That's what Schlomo Staszewski and Frankfurt colleagues tried to do in a study of 258 people taking TDF (21). Staszewski's group genotyped virus from everyone who didn't respond to a TDF regimen or had a viral rebound while taking the drug. The lion's share of cohort members—87%—had taken a median of seven antiretrovirals before starting TDF. When follow-up in this analysis began, they had a median viral load of 1330 copies/mL and a median CD4 count of 234 cells/µL.
After a median follow-up of 95 weeks, none of the 37 people taking TDF as part of their first regimen had K65R. Among the 258 people who started TDF with antiretroviral experience, K65R emerged in 29 (11%). The mutation cropped up most often in people taking TDF plus ddI or abacavir, usually with other nukes:
• 13 of 35 (37.1%) taking TDF + ddI + other nucleosides
• 13 of 61 (21.3%) taking TDF + abacavir + other nucleosides
• 3 of 24 (12.5%) taking TDF + ddI + a nonnucleoside
• 2 of 54 (3.7%) taking TDF + a nonnucleoside (without ddI or abacavir)
• 1 of 28 (3.6%) taking TDF + ddI + a ritonavir-boosted PI
The mutation emerged in none of 17 people taking TDF with abacavir and a ritonavir-boosted PI, none of 13 taking TDF with other nukes excluding ddI and abacavir, and none of 39 taking TDF and a ritonavir-boosted PI without ddI or abacavir.
A multivariate analysis singled out one factor that independently raised the risk of K65R:
• Taking TDF only with nucleosides raised the risk 8.5 times (P = 0.004)
Two factors independently lowered the risk of K65R:
• Taking AZT with TDF lowered the risk 93% (P = 0.024)
• Taking 3TC with TDF lowered the risk 85% (P = 0.016)
This keen interest in K65R is only one sign of the upheaval in thinking on resistance to nucleosides and --tides over the past few years. Gilead's Michael Miller rendered a service to the resistance-wary by posting a timely scorecard at the top of his report on K65R and L74V, a mutation seen with ddI or abacavir (22):
1. TDF can select K65R.
2. Abacavir or ddI can also select K65R along with L74V/I or mixtures thereof.
3. But abacavir typically selects M184V/I before K65R or L74V/I.
4. 3TC and FTC also select M184V.
5. AZT and d4T select the so-called thymidine analog mutations (TAMs): M41L, E44D, D67N, K70R, V118I, L210W, T215Y/F, K219Q/E (5).
6. M184V/I increases susceptibility to AZT, d4T, and TDF but decreases susceptibility to 3TC, abacavir, and FTC.
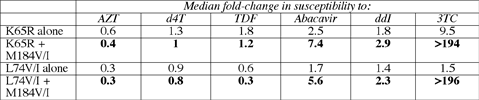
He could have added, as Staszewski's study and others show, that AZT stifles emergence of K65R. Cataloging mutations in more than 16,000 viral isolates sent to ViroLogic for resistance testing since early 2003 and comparing results from his earlier research, Miller traced an apparent plateau in emergence of K65R beginning around the second quarter of 2003. Prevalence of the mutation rose from 1.6% in the fourth quarter of 2001 to 2.3% in the second quarter of 2002, to 3.7% in the fourth quarter of 2002, and to 4.8% in the second quarter of 2003. In the following three quarters the prevalence of K65R hovered between 4.0% and 4.3%. Focusing just on 432 recently collected K65Rs and 1120 L74V/Is, Miller found that those mutations appear together most often with the 3TC-evoked M184V/I. But otherwise K65R and L74V/I tend to consort with different companions. L74V/I mingles most often with TAMs (see point 5 above), while K65R hangs out with an odd lot including S68G, A62V (part of the position 69 multinuke-resistance complex), and Y115F (linked to abacavir). When teaming up with either K65R or L74V/I, the M184V/I mutation yielded almost identical resistance profiles for all the drugs studied except TDF:
|
|
| |
| |
 |
|
| |
| |
The L74V/I mutation—either alone or with M184V/I—remained susceptible to TDF in this cell culture analysis. But Miller observed that the story may be different in people with this mutation. Earlier work by his group showed that K65R tended to emerge in people who pick up L74V/I with ddI or abacavir, and together those mutations limited TDF activity (23).
Mark Mascolini writes about HIV infection (mailmark@ptd.net).
References
1. Little SJ, Holte S, Routy JP, et al. Antiretroviral-drug resistance among patients recently infected with HIV. N Engl J Med 2002;347:385-394.
2. Little SJ, Routy JP, Collier AC, et al. The spectrum and frequency of reduced antiretroviral drug susceptibility with primary HIV infection in North America. Antiviral Ther 2000;5(suppl 3):134. Abstract 172.
3. Weinstock HS, Zaidi I, Heneine W, et al. The epidemiology of antiretroviral drug resistance among drug-naive HIV-1-infected persons in 10 US cities. J Infect Dis 2004;189:2174-2180.
4. Ross LL, Lim ML, Liao Q, et al. Prevalence of antiretroviral drug resistance and resistance mutations in antiretroviral therapy naive HIV infected individuals from 40 US cities during 2003. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). October 30-November 2, 2004. Washington, DC. Abstract H-173.
5. Johnson VA, Brun-Vézinet F, Clotet B, et al. Update of the drug resistance mutations of HIV-1: 2004. Topics HIV Med 2004;12:119-124. http://www.iasusa.org/resistance_mutations/mutations_figures.pdf.
6. Nettles RE, Kiefer T, Kwon P, et al. Intermittent low-level HIV-1 viremia in patients on HAART: defining blip dynamics, clinical correlates and genotypic resistance implications by frequent sampling and clonal ultrasensitive genotyping. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). October 30-November 2, 2004. Washington, DC. Abstract H-1134.
7. Havlir DV, Bassett R, Levitan D, et al. Prevalence and predictive value of intermittent viremia with combination HIV therapy. JAMA 2001;286:171-179.
8. Mira JA, Macias J, Nogales C, et al. Transient rebounds of low-level viraemia among HIV-infected patients under HAART are not associated with virological or immunological failure. Antivir Ther 2002;7:251-256.
9. Sklar PA, Ward DJ, Baker RK, et al. Prevalence and clinical correlates of HIV viremia ('blips') in patients with previous suppression below the limits of quantification. AIDS 2002;16:2035-2041.
10. Greub G, Cozzi-Lepri A, Ledergerber B, et al. Intermittent and sustained low-level HIV viral rebound in patients receiving potent antiretroviral therapy. AIDS 2002;16:1967-1969.
11. Raboud JM, Rae S, Woods R, et al. Consecutive rebounds in plasma viral load are associated with virological failure at 52 weeks among HIV-infected patients. AIDS 2002;16:1627-1632.
12. Delaugerre C, Teglas JP, Treluyer JM, et al. Predictive factors of virologic success in HIV-1-infected children treated with lopinavir/ritonavir. JAIDS 2004;37:1269-75.
13. Kantor R, Fessel WJ, Zolopa AR, et al. Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy. Antimicrob Agents Chemother 2002;46:1086-92.
14. Edwards D, Stalzer B, Napravnik S, et al. HIV resistance evolution in the setting of stable antiretroviral therapy. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). October 30-November 2, 2004. Washington, DC. Abstract H-176.
15. Parikh U, Koontz D, Hammond J, et al. K65R: a multi-nucleoside resistance mutation of low but increasing frequency. XII International HIV Drug Resistance Workshop. June 10-14, 2003. Los Cabos, Mexico. Abstract 136.
16. Winston A, Pozniak A, Mandalia S, et al. Which nucleoside and nucleotide backbone combinations select for the K65R mutation in HIV-1 reverse transcriptase? AIDS 2004;18:949-951.
17. Gallant JE, Rodriguez AE, Weinberg W, et al. Early non-response to tenofovir + abacavir (ABC) and lamivudine (3TC) in a randomized trial compared to efavirenz + ABC and 3TC: ESS30009 unplanned interim analysis. 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy. September 14-17, 2003. Chicago. Abstract H-1722a.
18. Farthing C, Khanlou H, Yeh V. Early virologic failure in a pilot study evaluating the efficacy of abacavir, lamivudine and tenofovir in the treatment of naive HIV-infected patients. 2nd IAS Conference on HIV Pathogenesis and Treatment. July 13-16, 2003. Paris. Abstract 43.
19. Landman R, Peytavin G, Descamps D, et al. Low genetic barrier to resistance is a possible cause of early virologic failure in once-daily regimen of abacavir, lamivudine, and tenofovir: the Tonus study. 11th Conference on Retroviruses and Opportunistic Infections. February 8-11, 2004. San Francisco. Abstract 52.
20. Jemsek J, Hutcherson P, Harper E. Poor virologic responses and early emergence of resistance in treatment naive, HIV-infected patients receiving a once daily triple nucleoside regimen of didanosine, lamivudine, and tenofovir DF. 11th Conference on Retroviruses and Opportunistic Infections, San Francisco, 2004. Abstract 51.
21. Staszewski S, Dauer B, Stuermer M, et al. Predictors of K65R development with tenofovir DF (TDF)-containing regimens in HIV therapy-experienced patients. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). October 30-November 2, 2004. Washington, DC. Abstract H-177.
22. McColl DJ, Parkin NT, Miller MD. Characterization of patient-derived HIV-1 isolates containing the L74V or K65R mutations in reverse transcriptase. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). October 30-November 2, 2004. Washington, DC. Abstract H-178.
23. Bae AS, Waters JM, Margot NA, et al. Pre-existing L74V is a risk factor for virological non-response after development of K65R in patients taking tenofovir. XIII International HIV Drug Resistance Workshop. June 8-12, 2004. Tenerife. Abstract 158.
|
| |
|
|
|
|
|