Reported by Jules Levin
"Final Results of a Phase I/II Dose Escalation Trial of Valtorcitabine LdC) in Patients with Chronic Hepatitis B"
S.G. Lim1, C.L. Lai2, M. Myers3, M.F. Yuen2, C.T. Wai1, D. Lloyd3, K. Pietropaolo3, X.J. Zhou3, G. Chao3, N.A. Brown3
1 Department of Medicine, National University Hospital, Singapore, Singapore
2 University Department of Medicine, University of Hong Kong, Hong Kong, China
3 Idenix Pharmaceuticals, Cambridge, MA, USA
Lim presented this information at the 40th EASL Meeting in Paris, April 13-17, 2005.
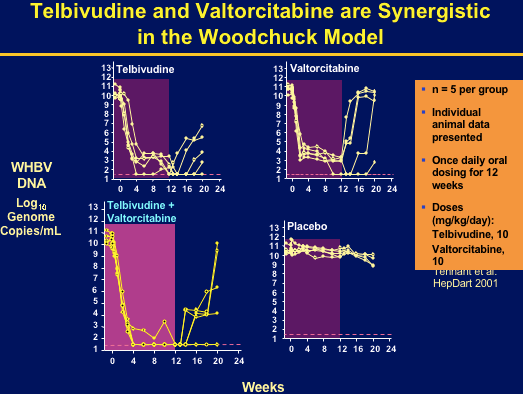
There is a continuing need for more effective therapies for chronic hepatitis B (CHB). L-deoxycytidine (LdC) and L-thymidine (telbivudine; LdT) are potent inhibitors of hepatitis B virus (HBV) replication in vitro. A one-year phase IIb trial demonstrated significantly greater viral suppression and serum ALT normalization in CHB patients receiving telbivudine, compared with lamivudine (Lai, AASLD2004). Valtorcitabine, a well-absorbed prodrug of LdC, is synergistic with telbivudine for inhibiting HBV replication in vitro and in the woodchuck hepadnavirus model. Valtorcitabine has been evaluated in CHB patients as the first step towards the potential development of an effective combination therapy with telbivudine.
The antiviral efficacy, safety, and pharmacokinetics of two valyl ester prodrugs of LdC were evaluated in a phase I/II dose escalation trial. First, a 3¢,5¢-divalyl LdC prodrug was investigated in sequential dose cohorts of 50, 100, 200, and 400 mg/day. Subsequent cohorts (300, 600, 900, and 1200 mg/day) investigated the more stable 3¢-monovalyl form of valtorcitabine. The 300 mg dose of monovalyl-LdC and the 400 mg dose of divalyl-LdC contain similar amounts of the LdC parent nucleoside and provided similar systemic LdC exposure. Each cohort comprised 7 HBeAg+ CHB patients, randomized 6:1 (drug vs. placebo). Serum HBV DNA >10 million (107); serum ALT <5 x ULN. 2 clinical sites: Singapore, Hong Kong). Patients were evaluated weekly during 28 days of treatment, with 12 weeks of follow-up, for serum HBV DNA levels and safety. Emax modeling was used to assess the quantitative relationship between dose and antiviral response at Week 4.
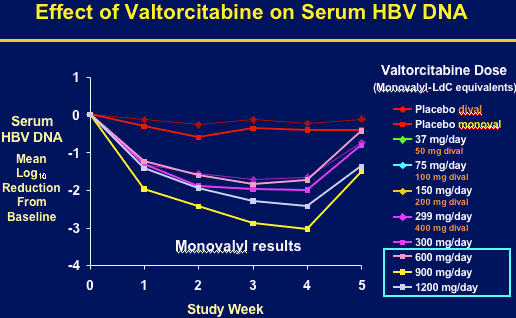
Consistent, dose-related HBV DNA reductions were observed, ranging from a mean 1.63 log10 for the 50 mg/day group at Day 28, to 3.04 log10 copies/ml at 900 mg/day. Emax modeling confirmed a progressive increase in HBV DNA suppression toward the theoretical maximum at doses up to 900 mg/day, with a diminished effect thereafter. Safety appeared comparable to placebo, with no treatment-related pattern of adverse events or laboratory abnormalities. 2 post-treatment ALT flares (both resiolved).
Valtorcitabine demonstrated substantial suppression of serum HBV DNA and was well tolerated in patients with chronic hepatitis B. A dose of 900 mg/day maximized viral suppression with mean 3.04 log reduction (99.9%) at week 4. The presenter said viral suppression was proportionately greater than that reported for lamivudine, adefovir, about the same as entecavir, but less than telbivudine. 900 mg dose was selected for ongoing clinical evaluation of valtorcitabine in combination with telbivudine.
This study supports a "proof-of-concept" phase IIbv trial of combination therapy (valtorcitabine+telbivudine), now underway:
Telbivudine 600mg/day vs telbivudine 600 + valtorcitabine 900 mg/day; 52 weeks treatment; compensated HBeAg+ patients with high viral load. The goal is to increase the percent of PCR-negative by 6 months.
Idenix presented in a poster the results of a drug interaction study between LdT & two HBV drugs:
"Absence of Pharmacokinetic Drug-Drug Interaction Between Telbivudine and Lamivudine or Adefovir Dipivoxil in Healthy Subjects"
The effect of lamivudine (100 mg/d) on telbivudine (200 mg/d)
pharmacokinetics was a slight (<8%) increase in Cmax, with no effect on AUCτ. The effect of telbivudine on lamivudine pharmacokinetics was a decrease in Cmax (geometric mean ratio of 86.0%), with no effect on AUCτ. Although the magnitudes of these effects were not large and therefore unlikely to be clinically relevant, the 90% CIs were outside the target range of 80% to 125% for Cmax. The 90% CIs for AUCτ were within the target range for both drugs.
There were no statistically or clinically significant interactions between
telbivudine and lamivudine, or telbivudine and adefovir dipivoxil, with respect to the pharmacokinetic parameters of each drug.
Although the telbivudine dosage used to assess pharmacokinetics with
lamivudine was 200 mg and lower than the intended clinical dose of 600 mg, clinically significant interactions between lamivudine and telbivudine at 600 mg are unlikely, especially in light of the lack of effect on AUC with combination therapy.
Telbivudine alone or in combination with lamivudine or adefovir dipivoxil appeared to be safe and well tolerated in healthy subjects.
SAFETY
Lamivudine Interaction Study
A total of 28 treatment-emergent AEs were reported by 11 (69%) of the 16 subjects dosed. Sixteen AEs were reported in group 1 subjects and 12 AEs were reported in group 2 subjects.
Arthralgia, dizziness, and headache were the most common AEs reported. No serious AEs occurred during the study, and no subjects were discontinued from the trial because of AEs.
There were no treatment-related trends observed regarding clinical laboratory parameters, vital sign measurements, or physical examination findings.
Adefovir Dipivoxil Interaction Study
A total of 24 AEs were reported by 11 (69%) of the 16 subjects during the trial. Seven group 1 subjects reported 12 AEs and four group 2 subjects reported 12 AEs. Most AEs were reported as mild in intensity (92%) and were considered to be reasonably or possibly related to the study drug (79%).
Headache was the AE reported by the greatest number of subjects, followed by nausea. There were no serious AEs reported in this study, and no subjects were discontinued because of AEs.
No clinically significant changes from baseline were noted in clinical
laboratory results or vital sign assessments following dosing.
Hepatitis B Therapeutic Goals
Profound & sustained viral suppression
Minimal viral resistance
Excellent tolerability, even with extended therapy
Once daily oral dosing for maximal adherence
Improved rate of clinically relevant efficacy outcomes:
--HBeAg & HBsAg seroconversion
--Improved liver histology
--Improved long-term outcomes
Combination therapy may optimize sustained viral suppression, particularly for dofficult to treat patients. Note from Jules Levin: so far, clinical studies have not established efficacy of combination therapy, except for the finding that combination therapy reduced the development of resistance. This, in and of itself, would be a worthwhile accomplshment.
HBV Specific L-Nucleosides: Valtorciabine (val-LdC) and Telbivusine (LdT)
Small molecule HBV polymerase inhibitors.
HBV-specific.
High intracellular triphosphate concentrations.
Once daily oral dosing.
Favorable toxicology.
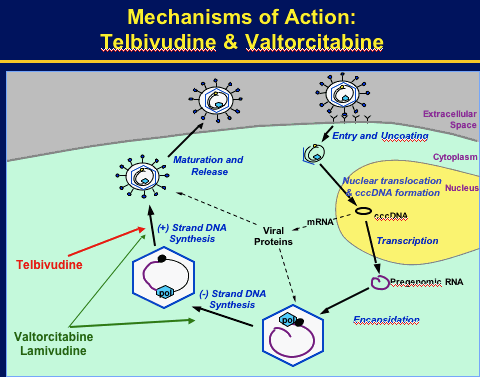
Potentially complementary mechanisms of action on 1st & 2nd strand HBV DNA synthesis.
Combination has potent synergistic antiviral activity in vitro & ib woodchuck HBV model.
Both compounds have excellent safety profiles.
Hepatitis B Clinical Program
Telbivudine phase II clinical trials ongoing.
Investigate telbivudine + valtorcitabine as combination for difficult to treat patients, after monotherapy data become available for both agents.
Baseline Characteristics
All patients were Asian & HBsAg & HBeAg+ at baseline.
HBV DNA (median log c/ml)
Divalyl LdC (mg/day)
8.8 (median log copies/ml) placebo
8.7 - 50 mg/day
9.3 - 100mg
9.1 - 200mg
7.1 - 400mg
Monovalyl LdC (mg/day)
9.1 — placebo
9.0 — 300mg
7.9 — 600mg
9.1 - 900mg
8.7 — 1200mg
Potentially complementary mechanisms of action on 1st & 2nd strand HBV DNA synthesis.