 |
|
|
| |
European Resistance Workshop-Part 1. Tenofovir, Nucleosides, and Nonnucleosides (TMC278, NVP in MTCT, ddI/TDF/EFV viral failure)
|
To view slides from oral presentations and most posters from the Third European HIV Drug Resistance Workshop, go to
http://www.hivpresentation.com.
|
| |
 |
| |
Despite a grand and growing panoply of antiretrovirals, how little we know about HIV can be humbling.
For example, noted Hans-Georg Kraeusslich (University Clinic, Heidelberg), several schematic slides at the Third European HIV Drug Resistance Workshop portrayed HIV as a crisply uniform icosahedron-a 20-sided globe [1]. His own immaculate slides of ice-coated virions showed snowball-smooth spheres of discrepant size.
But even Kraeusslich's slides lie, he freely admitted. A trick-animation look at viral budding and maturation-pasted together from a series of still photos-suggested that HIV's core protein condenses from airy nothingness inside the viral shell, just as Marley's ghost materializes to haunt Scrooge. But Kraeusslich's research shows that the core builds up bit by bit from one end to the other.
Questions of more clinical urgency haunt all HIV clinicians trying to treat people with drug-resistant virus:
-- Will zidovudine (AZT) heed recent theory and constrain the K65R mutants oft provoked by tenofovir disoproxil fumarate (TDF)?
-- Do proliferating reports of rapid resistance to enfuvirtide (T-20) point to limits with this fusion inhibitor-or only to limited skill in its use?
-- Will transmission of resistant virus rise unabated-as in some European countries-or settle onto a plateau-as in others?
-- Will tipranavir emerge as a muscular alternative for people with protease inhibitor-resistant virus, or simply as another stopgap after multiple regimen failures?
-- Will the next likely antiretroviral class, the CCR5 antagonists, prove a powerful new weapon-or will they spur HIV to fashion faster routes to progression?
Staring into the face of HIV for 20-odd years, clinicians and researchers have had little more than a blank stare back. The face of the retrovirus sometimes seems as nearly inscrutable as the face of those enigmatic Cycladic carvings one could see a few blocks down the street from the Athens Hilton, site of the March 30-April 1 Third European Drug Resistance Workshop.
There, in the Museum of Cycladic Art, wandering workshop attendees may have viewed these peculiar pocket-sized sculptures, whose very meaning remains unknown. Guides to the next life? Gravestead companions? Toys?
These nearly uniform icons-arms clenched over gut in classic defensive stance-offer little hint. Even their lovely lyre-shaped heads have to little to say; lacking mouths, eyes, and usually ears, they apprehend would-be interlocutors only olfactorily, with a triangular nose sloping up from a flat face.
Like HIV, though, these so-called canonical figurines had not 20 faces, but uncounted aspects. Time has rubbed off their facial eccentricities, once painted on by Cycladic crafters. These myriad physiognomies appear now only as the faintest tinctures on larger figures. HIV may similarly seem a many-faced blank slate for clinicians trying to freeze its always evolving features with genotype or phenotype. Yet researchers developed a few more frames at the European Resistance Workshop, detailed below and in forthcoming reports on the meeting.
This installment covers studies on resistance to nucleotides, nucleosides, and nonnucleosides. Subsequent segments explore workshop reports on resistance to protease inhibitors, resistance to entry inhibitors, evolution of resistance during multiregimen failure, pharmacokinetics and resistance, transmission of resistant virus, and resistance among HIV-1 subtypes.
Tenofovir resistance and rescue: more on K65R
K65R can be bad news. Its emergence, often linked to tenofovir disoproxil fumarate (TDF), can compromise viral susceptibility to all marketed (and many experimental) nucleosides-with one intriguing exception: AZT [2]. A possible explanation for this variance emerged in work by Urvi Parikh in the University of Pittsburgh lab of John Mellors [3]. K65R apparently remodels reverse transcriptase in a way that prevents excision of AZT-and so stops resistance to this drug through emergence of thymidine analog mutations (TAMs, M41L, D67N, K70R, L210W, T215Y/F, K219Q/E).
Mellors and Parikh theorize that giving AZT with other nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) can stifle evolution of K65R,[2] and they cite work buttressing this theory [4-6]. But can AZT beat back virus already tagged with K65R?
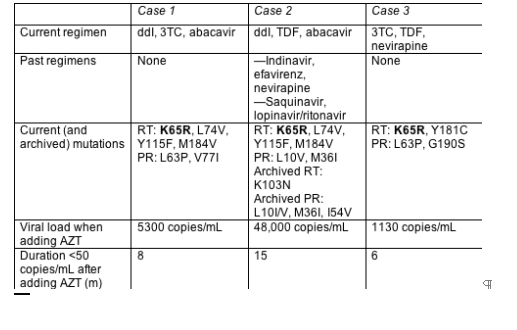
Yes, at least in three cases with K65R and no TAMs reported at the European Resistance Workshop by Schlomo Staszewski (J.W. Goethe University Hospital, Frankfurt).[7] All three people added AZT to failing regimens without switching the other drugs and enjoyed speedy and prolonged visits to sub-50-copy territory. The three shared two key traits-a detectable viral load while taking a regimen that provoked K65R, and no TAMs (Table 1.1).
|
|
 |
| |
Baseline mutation patterns suggested resistance to all three drugs in the failing regimen. Yet in all three people the viral load plunged immediately after they added AZT and has remained undetectable for 6 to 15 months. CD4 counts stayed stable (in cases 1 and 3) or climbed (in case 2).
What can one make of three case reports? Not a universal intensification strategy, to be sure. The reports do endorse the idea that K65R-tainted virus remains susceptible-perhaps highly susceptible-to AZT. And they may provide tinder for controlled trials.
But the oblique interface between K65R and TAMs needs more study. Recent work by Pittsburgh's Parikh determined that K65R and the keystone TAM T215Y/F/I never sit on the same genome, even when population sequencing detects both in plasma samples [8]. But clonal analysis by a French team found that the same viral clone can bear K65R and an array of TAMs [9].
A chart review by clinicians at two Spanish hospitals, Ramn y Cajal in Madrid and Germans Trias i Pujol in Badalona, confirmed Staszewski's finding that K65R mutants can respond to regimens that still include TDF, didanosine (ddI), or abacavir [10]. In a separate, larger study, Carolina Gutierrez (Ramn y Cajal Hospital) rated K65R much more likely after failure with TDF than with ddI or abacavir [11].
The two-hospital enquiry analyzed K65R trends in 33 people treated from 1996 to 2004. The mutation arose more often with TDF (n = 21) than with ddI (n = 17) or abacavir (n = 11). A lamivudine (3TC)-linked M184V mutation popped up in 11 people taking TDF (52%), 9 taking ddI (54%), and 7 taking abacavir (54%).
After K65R emerged, 16 people started a regimen containing ddI, 12 a combination including TDF, and 8 an abacavir regimen. Virologic response after a median 12 weeks of follow-up (range 2 to 60 weeks) did not differ between those starting ddI (-1.1 log), TDF (-1.0 log), or abacavir (-1.3 log). And the RNA decline rate proved similar with or without M184V. Gutierrez did not report how many people took AZT after K65R appeared, so the impact of that nucleoside cannot be assessed here.
In the single-hospital study, Gutierrez sequenced 981 viral isolates and found K65R in 21 (2%). That rate vaulted from 0.2% in 842 isolates collected in 1996-2002 to 13.7% in 139 isolates from 2003-2004 (P < 0.001). K65R arose with 13 of 97 TDF regimen failures (13%) compared with 9 of 153 abacavir failures (6%) and 9 of 344 ddI failures (3%) (P < 0.001). Regardless of which of these three NRTIs people took, K65R emerged significantly more often with triple-NRTI regimens than with NRTIs plus a nonnucleoside or protease inhibitor (P = 0.001).
Another look at ddI/TDF/efavirenz failure
High rates of early virologic failure with regimens combining ddI and TDF with a nonnucleoside-usually efavirenz [12-14]-prompted a letter from Gilead and Bristol warning European clinicians to avoid this NRTI doubling, especially in people starting treatment with a high viral load [15]. An Italian trial that randomized treatment-naive people to ddI/TDF/efavirenz, 3TC/TDF/efavirenz, or AZT/3TC/lopinavir/ritonavir shed more light on viral vicissitudes with ddI/TDF [16].
Despite the small size of this 30-person pilot by Carlo Torti (Institute for Infectious and Tropical Diseases, Brescia), it does merit interest because of the once-daily comparison of ddI versus 3TC with TDF/efavirenz and because of an intriguing pharmacokinetic hint. Although pretreatment traits did not differ significantly between study arms, the ddI/TDF group began with the lowest median CD4 count (113 cells/µL, interquartile range 42 to 231 cells/µL) and the highest median load (4.78 logs-about 60,250 copies/mL-IQR 4.36 to 5.25 logs).
After 14 weeks of treatment 6 of 9 people (67%) taking AZT/3TC/lopinavir, 9 of 10 (90%) taking 3TC/TDF/efavirenz, and 5 of 11 (45%) taking ddI/TDF/efavirenz had fewer than 50 copies/mL. After 28 weeks of therapy 7 of 8 (87.5%) on AZT/3TC/lopinavir, all 10 on 3TC/TDF/efavirenz, and 6 of 10 on ddI/TDF/efavirenz were under the 50-copy mark. Plasma loads dropped significantly faster with the 3TC/TDF combo than with ddI/TDF (P = 0.0001).
In 3 people who failed to lower their viral load at least 10-fold by week 4 with ddI/TDF/efavirenz, the nonnucleoside mutations K103N, Y188L, and G190E emerged first, followed by K65R. The nucleoside-educed D67N and K219Q substitutions arose in one person and L210F plus T215D in another.
Efavirenz levels measured over 24 hours on day 7 proved substantially lower with early failure of ddI/TDF/efavirenz (22.29 h o mg/L) than in people who responded to that regimen (49.10 h o mg/L).
New wrinkles in resistance to TDF?
Two workshop studies tried to fine-tune resistance analyses of TDF and ddI. Federico Garcia (Hospital San Cecilio, Granada) and colleagues at other clinics divined the impact of mutations that improve the response to these two drugs [17], while Chris Stone (GlaxoSmithKline) proposed new TDF-linked mutations [18].
Garcia's study involved 161 people taking TDF and 139 taking ddI. More than 80% in each group had at least two virologic failures on their charts, and median numbers of reverse transcriptase and protease mutations topped 4 in both groups [17]. The TDF cohort had a median viral load just above 10,000 copies/mL and the ddI group just below that mark.
To devise genotypic scores, Garcia started with all mutations on the International AIDS Society (IAS)-USA list and selected those culled at a 10% or greater rate and tied to a 3-month change in viral load (at P < 0.2) when starting a new regimen.
For TDF the most predictive model included six mutations linked to a worse virologic response and one linked to a better response. For ddI four mutations presaged a worse response and two a better response:
-- "Bad" mutations for TDF: Changes at reverse transcriptase positions 41, 44, 67, 118, and 210, and T215Y
-- "Good" mutations for TDF: Changes at reverse transcriptase position 184
-- "Bad" mutations for ddI: Changes at reverse transcriptase positions 67, 118, and 210, and T215Y
-- "Good" mutations for ddI: Changes at positions 70 and 184
For both drugs, a genotypic score that included only the "bad" mutations correlated well with the 3-month change in viral load. But subtracting from the resistance scores for each "good" mutation yielded tighter correlations with more highly significant P values. (A higher r value means a better correlation.)
-- Pearson r for TDF "bad" mutation score 0.242 (P = 0.002)
-- Pearson r for TDF "bad" minus "good" mutation score 0.284 (P = 0.0002)
-- Pearson r for ddI "bad" mutation score 0.307 (P = 0.00024)
-- Pearson r for ddI "bad" minus "good" mutation score 0.332 (P = 0.000064)
Although the good-minus-bad score may offer a more precise assessment of response to TDF and ddI, one workshop attendee observed, Garcia's TDF model may not be the best response predictor because it lacks the K65R mutation. That seeming anomaly arose because Garcia and colleagues found K65R at a rate below 10% in their population, so they could not include it in the model.
In vitro serial passage studies by Glaxo's Chris Stone linked the Y115F reverse transcriptase mutation to TDF for the first time and found that TDF plus emtricitabine (FTC) and 3TC/abacavir/TDF first select K65R, even though FTC and 3TC strongly promote selection of M184V or M184I [18].
Serial passage studies expose HIV-infected lab cell lines (in this case HXB2 cells) to increasing concentrations of one or more drugs with each passage. The goals are to see how much drug pressure it takes to evoke mutations and which mutations arise in which sequence.
In a serial passage study involving TDF alone, the drug selected K65R in the fourth passage. In the eight passage (representing a 32-fold increase in the concentration needed to inhibit nonmutant virus at a 50% rate), Y115F popped up. Stone observed that this appears to be the first time TDF alone selected Y115F, a mutation that confers resistance to abacavir but is not tied to other NRTIs.
Abacavir plus 3TC selected M184I or M184V after a single passage, as expected. But TDF/FTC first selected two mutations-K65R and V10I-at passage two, even though FTC alone mirrors 3TC in strongly selecting M184V/I. The preference for K65R over M184V/I with TDF/FTC, Stone surmised, may reflect M184V's tendency to make virus more susceptible to TDF. Thus TDF pressure would militate against its emergence. The import of V10I in this experiment remains a mystery. No earlier evidence links that substitution to resistance.
After two passages 3TC/abacavir/TDF selected K65R. After seven passages Y115F appeared.
Stone cautioned that lab results like these don't always match what happens in people who take the drugs. For example, in human studies both TDF/FTC and 3TC/abacavir/TDF select M184V/I, whereas that mutation did not arise in Stone's serial passage studies of those combinations. One likely reason for this difference is that passage studies use a single HIV strain, while infected people almost always harbor a diverse population of major and minor strains when they start therapy. Still, Stone suggested further research to define a possible tie between TDF and Y115F.
Tracing divergent TAM pathways
Thymidine analog mutations (TAMs) summoned by AZT or stavudine (d4T) tend to travel along one of two pathways-and which path they take affects cross-resistance to NRTIs. Key cobblestones along the TAM-1 path (M41L, L210W, and T215Y) pave a faster route to cross-resistance than the TAM-2 markers D67N, K70R, T215Y, and K219E/Q.
But what factors favor one TAM path over the other? To find out, Andrea De Luca (Catholic University, Rome) sifted mutations and other variables in 1464 people with TAMs and at least 3 months of potent combination experience [19].These were heavily treated people, with an average 67 months of NRTI experience and an average 32 months on potent therapy. Two thirds tried NRTI monotherapy and 81% sampled dual NRTI therapy. The number of regimens taken averaged six and the number of antiretrovirals seven.
In his search for pathway predictors, De Luca used precise definitions of TAM-1, TAM-2, and TAM-3 (neither 1 nor 2):
-- TAM-1: Either one TAM from this set of three-M41L, L210W, and T215Y-or more
than one TAM with virus nonmutant at position 70 plus either L41L or L210W or both
-- TAM-2: Either one TAM from this set of five-D67N, K70R, T215F, and K219E/Q-or more than one TAM with virus nonmutant at positions 41 and 210
-- TAM-3: Neither of the above patterns
In this cohort HIV preferred the TAM-2 route before 1996, at a prevalence of 72%. That rate plunged to 30% by 2000. Meanwhile, TAM-1 prevalence leapt from 20% before 1996 to 59% in 2001, then eased to 53% in 2003-2004. Throughout this period the TAM-3 rate hovered between 8% and 15%.
In a multivariate analysis several factors predicted a higher risk of the more dangerous TAM-1 route: d4T/3TC in the last potent combination, more regimens in the treatment history, and experience with nevirapine, efavirenz, or ritonavir. A longer time on NRTI monotherapy lowered the risk of TAM-1 mutations.
On the other hand, several of these same factors independently favored a lower risk of the TAM-2 route: d4T/3TC in the last regimen, more regimens, and longer duration of nonnucleoside therapy. Amprenavir experience also lowered the odds of collecting TAM-2 mutations.
Analysis of which reverse transcriptase substitutions cluster with TAM-1 or TAM-2 mutations suggested that several unheralded mutations contribute to still-recondite resistance dynamics. Valentina Svicher (University of Rome Tor Vergata) and colleagues at other centers inched their way through this dim maze by figuring correlation coefficients for any pair of mutations spotted in 551 treatment-naive and 1355 NRTI-treated people [20].
This exercise reaped a dozen little-remarked reverse transcriptase substitutions linked to NRTI therapy: K20R, V35M, T39A, K43E/N/Q, K122E, G196E, E203D/K, H208Y, and D218E. The natural polymorphisms R83K and I50V routinely cropped up in untreated people, but rarely in those taking NRTIs. The R83K switch, Svicher found, almost never clustered with the TAM-2 mutations D67N, K70R, and K219Q (frequency 0.4%, P < 0.001), while I50V resolutely shunned M184V (frequency 1%, P = 0.15). She proposed that these substitutions pump up the genetic barrier to NRTI resistance.
The common F214L polymorphism also rarely appeared with TAM-2 mutations (frequency 4.5%, P < 0.001), but it avoided the TAM-1 mutations M41L, L210W, and T215Y even more doggedly (frequency 0.07%, P < 0.001). On the other hand, five substitutions clustered with the TAM-1 group: K43E/Q, K122E, G196E, and H208Y. But the D218E flip preferred TAM-2 mutations.
Svicher suggested that the substitutions she identified should be considered in algorithms used to translate genotypes into resistance scores.
Resistance to TMC278, a new nonnucleoside
Tibotec grooms two novel nonnucleosides in its stable of antiretroviral hopefuls, TMC125 and TMC278. Though TMC125 has seniority, recent reports fattened the resume of TMC278. A dose-finding study unveiled at this year's Conference on Retroviruses charted a 1.2-log viral load drop in 7 days with TMC278, regardless of the dose used [21]. At the European Resistance Workshop, Tibotec's Marie-Pierre de Bethune offered a resistance analysis of the drug [22].
Tested against 10 strains bearing one or two nonnucleoside mutations, TMC278 retained a 50% effective concentration (EC50) below 1 nM against five of them (L100I, K103N, V106A, G190A, G190S) and below 2.8 nM for the others (K101E, Y181C, Y188L, L100I/K103N, K103N/Y181C). Efavirenz, on the other hand, had an EC50 above 10 nM against eight of these 10.
In a test against more than 1500 recombinant nonnucleoside-resistant clinical isolates, TMC278 retained full activity (EC50 below 1 nM) against nearly 60% and some activity (EC50 below 10 nM) against most of the rest. Nearly every recombinant virus in this panel proved resistant to nevirapine, and about 40% were highly resistant to efavirenz.
Whereas 1 µM of nevirapine or efavirenz promptly selected resistant virus in high-multiplicity-of-infection experiments, neither 1 nor 40 µM of TMC278 selected resistant virus through 32 days of study. While only one mutation can render nevirapine or efavirenz impotent, eight mutations (including L100I, V106I, Y181C, and M230I) piled up before virus lost susceptibility to TMC278.
Though Bethune vowed that Tibotec has not dropped development of TMC125, TMC278 may have a pharmacologic edge-one-pill, once-daily dosing seems likely. Clinical trials of TMC125 used twice-a-day dosing.
Resistance after single-dose nevirapine
Single doses of nevirapine for a woman in labor and her newborn child are the simplest way to lower the risk of HIV transmission. But the strategy almost always evokes resistant virus, even when standard assays can't spot it. Laurence Vergne (Centre Muraz Unite VIH, Bobo Dioulasso, Burkina Faso) added another chapter to this sad chronicle with a study of 44 women in Burkina Faso and 37 in Cameroon [23].
The women carried a wide array of HIV-1 subtypes and circulating recombinant forms (CRFs), including 54% with CRF_02, 24% with CRF_06, 7% with subtype A, 4% with subtype G, and 3% each with CRF_01 or CRF_13. Vergne amplified reverse transcriptase in virus from all of the women in Cameroon and 79.5% of those in Burkina Faso.
The median time from nevirapine dosing to resistance testing measured 18 days in Burkina Faso and 24 days in Cameroon. Probably because of faster testing in Burkina Faso, Vergne recorded more resistant virus there-in 8 of 44 women (18%) versus 3 of 37 (8%) in Cameroon.
Two recent studies using assays that can see smaller traces of mutant virus charted resistance rates approaching 70% in South African women [24,25]. Both studies confirmed that highly sensitive PCR assays pick up mutations missed by standard population sequencing. Speaking at February's Conference on Retroviruses, resistance deans Douglas Richman and Franois Clavel voiced their belief that virtually everyone who takes single-dose nevirapine walks away with resistant virus. Even supersensitive techniques hunt for mutations only in plasma, whereas most resistant virus settles down in cells harder to assay.
Do these covert resistant strains pose a clinical threat? Yes, suggested two case studies detailed at the workshop by Marcelo Soares (Federal University of Rio de Janeiro, Brazil) [26]. He documented the K103N mutation in one child and K101E in another, although neither child had been exposed to nonnucleosides and standard genotyping did not spot the mutants in their mothers.
Analysis of 25 clones generated from the second mother's virus did turn up K101E in one clone. Twenty clones from the first mother showed no K103N, but standard genotyping saw that mutation in the father. Soares figured the father transmitted the virus to his wife, who still harbors the mutant even though clonal analysis missed it. Another possibility is that K103N arose spontaneously in the child.
References
To view slides and posters from the Third European HIV Drug Resistance Workshop, go to http://www.hivpresentation.com
1. Kraeusslich HG. HIV protease understanding-understanding protease resistance. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Invited lecture.
2. Parikh UM, Koontz DL, Chu CK, et al. In vitro activity of structurally diverse nucleoside analogs against human immunodeficiency virus type 1 with the K65R mutation in reverse transcriptase. Antimicrob Agents Chemother 2005;49:1139-1144.
3. Parikh U, Koontz U, Sluis-Cremer N, et al. K65R: a multinucleoside resistance mutation of increasing prevalence exhibits bi-directional phenotypic antagonism with TAM. 11th Conference on Retroviruses and Opportunistic Infections. February 8-11, 2004. San Francisco. Abstract 54.
4. Gulick RM, Ribaudo HJ, Shikuma CM, et al. Triple-nucleoside regimens versus efavirenz-containing regimens for the initial treatment of HIV-1 infection. N Engl J Med 2004;350:1850-1861.
5. Winston A, Mandalia S, Pillay D, et al. The prevalence and determinants of the K65R mutation in HIV-1 reverse transcriptase in tenofovir-naive patients. AIDS 2002;16:2087-2089.
6. Winston A, Pozniak A, Mandalia S, et al. Which nucleoside and nucleotide backbone combinations select for the K65R mutation in HIV-1 reverse transcriptase. AIDS 2004;18:949-957.
7. Staszewski S, Dauer B, Stuermer M, et al. Intensification of a failing regimen with AZT may cause sustained virologic suppression in the presence of the K65R mutation. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 89. Poster 9.34.
8. Parikh U, Barnas D, Bixby C, et al. K65R and T215Y are not present on the same viral genome in plasma samples with both mutations detected by population sequencing. 12th Conference on Retroviruses and Opportunistic Infections. February 22-25, 2005. Boston. Abstract 98.
9. Wirden M, Malet I, Derache A, et al. Clonal analyses of HIV quasispecies in patients harbouring plasma genotype with K65R mutation associated with thymidine analogue mutations or L74V substitution. AIDS 2005;19:630-632.
10. Gutierrez C, Perez-Elias MJ, Page C, et al. Virological response in HIV-1 infected patients with K65R mutation in the reverse transcriptase gene. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 87. Poster 9.32.
11. Gutierrez C, Moreno S, Perez-Elias MJ, et al. Selection of the K65R mutation in HIV-1 patients under different antiretroviral regimens. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 88. Poster 9.33.
12. Moyle G, Maitland D, Hand J, et al. Early virological failure in persons with viral loads >10,000 copies/ml and CD4 count <200 cells/mm3 receiving didanosine/tenofovir/efavirenz as initial therapy: 12 weeks results from a randomised controlled trial. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy. October 30-November 2, 2004. Washington, DC. Abstract H-566.
13. Podzamczer D, Ferrer E, Gatell JM, et al. Early virologic failure with a combination of tenofovir, didanosine and efavirenz. Antivir Ther 2005;10:171-177.
14. Leon A, Martinez E, Mallolas J, et al. Early virological failure in treatment-naive HIV-infected adults receiving didanosine and tenofovir plus efavirenz or nevirapine. AIDS 2005;19:213-215.
15. Carter M. 'Dear Dr' letter issued about risks of using tenofovir and ddI together. aidsmap.com. March 4, 2005. http://www.aidsmap.com/en/news/32A4DCAC-1FFD-4563-8919-81C5F25B455D.asp.
16. Torti C, Quiros-Roldan E, Regazzi M, et al. Factors associated with early virological failure after tenofovir + didanosine + efavirenz combination in naive patients: the KARINA-SISTHER group of the MASTER cohort. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 58. Poster 9.3.
17. Alvarez M, Carlos S, Palomares JC, et al. Effect of mutations with a positive impact on virological response over genotypic interpretation scores to tenofovir and didanosine. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 62. Poster 9.7.
18. Stone C, Craig C. Identification of a new HIV-1 in vitro mutation to tenofovir and mutation patterns observed to the drug combinations abacavir/tenofovir/3TC, abacavir/3TC and tenofovir/FTC. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 98. Poster 10.8.
19. De Luca A, Di Giambenedetto S, Romano L, et al. Prevalence and predictors of thymidine analogue mutations (TAM) patterns in HIV-1 from patients failing antiretroviral therapy: findings from the ARCA cohort study. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 59. Poster 9.4.
20. Svicher V, Ceccherini-Silberstein F, Sing T, et al. Additional mutations in HIV-1 reverse transcriptase are involved in the highly ordered regulation of NRTI resistance. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 63. Poster 9.8.
21. Goebel F, Yakovlev A, Pozniak A, et al. TMC278: potent anti-HIV activity in antiretroviral therapy-naive patients. 12th Conference on Retroviruses and Opportunistic Infections. February 22-25, 2005. Boston. Abstract 160.
22. de Bethune MP, Azijn H, Guillemont J, et al. In vitro characterization of TMC278, a novel potent NNRTI with an increased genetic barrier to the development of resistance. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 93. Poster 10.3
23. Vergne L, Kouankack C, Diagbouga S, et al. Selection of resistance mutations under nevirapine prophylaxis to prevent HIV-1 mother-to-child transmission in Africa. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 26. Poster 2.2.
24. Johnson J, Li JF, Morris L, et al. Resistance emerges in the majority of women provided intrapartum single-dose nevirapine. 12th Conference on Retroviruses and Opportunistic Infections. February 22-25, 2005. Boston. Abstract 100.
25. Palmer S, Boltz V, Maldarelli F, et al. Persistence of NNRTI-resistant variants after single-dose nevirapine in HIV-1 subtype-C-infected women. 12th Conference on Retroviruses and Opportunistic Infections. February 22-25, 2005. Boston. Abstract 101.
26. Afonso AO, Machado ES, Lambert JS, et al. Mother-to-child transmission of minority HIV-1 drug resistant strains. Third European HIV Drug Resistance Workshop. March 30-April 1, 2005. Athens. Abstract 25. Poster 2.1.
| |
| |
| |
| |
|
|
|
|
|