 |
|
|
| |
A PILOT 48-WEEK STUDY OF ONCE-DAILY (180 MG QD) VERSUS TWICE-DAILY (90 MG BID) ENFUVIRTIDE
|
| |
| |
Reported by Jules Levin
8th International Congress on Drug Therapy in HIV Infection
November 12-16, 2006, Glasgow
1Wright D, 2Rodriguez A, 3Godofsky E, 4Walmsley S, 5Labriola-Tompkins E,
5Shikhman A, 5Tucker E, 5Chiu Y-Y, 5Chung J,
5Rowell L, 6Graham N and 5Salgo M
1Central Texas Clinical Research, Austin, TX, USA; 2University of Miami, Miami,
FL, USA; 3Bach & Godofsky, Bradenton, FL, USA;
4University of Toronto, Toronto, ON, Canada; 5Roche, Nutley, NJ, USA; 6Trimeris, Morrisville, NC, USA
BACKGROUND
Enfuvirtide (ENF, FUZEON) is the only approved drug in the class of antiretrovirals (ARVs) known as HIV-1 fusion inhibitors.
Enfuvirtide is currently administered twice daily (bid) as a subcutaneous (SC) injection 12 hours apart, each injection delivering 90 mg of active drug. Once-daily (qd) dosing (180 mg enfuvirtide administered as 2 x 90 mg injections) would be expected to offer patients receiving enfuvirtide improved convenience.
The current formulation of enfuvirtide does not allow for 180 mg to be reconstituted in 1 mL of sterile water and a 1 mL injection volume is considered the maximum acceptable volume for the delivery of any SC therapeutic. Consequently, enfuvirtide at a dose of 180 mg qd must be administered as two injections once daily.
An earlier study, T20-104, demonstrated that short-term administration of enfuvirtide 180 mg qd compared to 90 mg bid resulted in bioequivalent exposure to enfuvirtide based on the area under the plasma concentration-time curve (AUC); however, the qd regimen was associated with a higher Cmax and lower Cmin.1 The impact of the lower Cmin on the efficacy of the qd regimen could not be determined based on the T20-104 study; therefore longer term studies are required to determine if 180 mg qd dosing is a safe and effective option for enfuvirtide administration.
The present study, T20-401, evaluated the efficacy and safety of enfuvirtide given as either 180 mg qd or 90 mg bid over 48 weeks of therapy.
AUTHOR DISCUSSION
This study assessed the efficacy of enfuvirtide over 48 weeks of therapy when dosed either once daily (given as 2 x 90 mg enfuvirtide qd) or as the standard twice-daily regimen (90 mg bid), both in combination with an OB regimen. The study was not powered to demonstrate non-inferiority of the qd dosing regimen.
After 48 weeks the percentage of patients reaching the primary endpoint of a viral load of < 400 copies/mL with enfuvirtide qd was similar to those achieving the same endpoint in the bid group. The proportion of patients achieving ≥ 1 log10 decrease from baseline was also similar across treatments, whereas a lower percentage of patients in the 180 mg qd group compared to the 90 mg bid group achieved a virological response of < 50 copies/mL.
The immunological benefit, as reflected by increases in CD4 count at 48 weeks, was comparable between the 180 mg qd group and the 90 mg bid group.
ISRs are commonly associated with enfuvirtide but in clinical trials rarely result in discontinuation of therapy.2 In this study, only one patient (in the qd arm) discontinued due to ISRs. Overall, similar frequencies of ISRs were detected in both the qd and the bid treatment arms. There was, however, a modest increase in the frequency of patients reporting more severe events in the qd arm compared to the bid arm.
As measured by 4-day recall, adherence to enfuvirtide was higher on the qd regimen compared to the bid regimen. Despite this, viral suppression was similar across the treatments, at least for the primary endpoint of < 400 copies/mL.
Subgroup analyses of those with ≥ 85% adherence suggested a modest trend towards better viral suppression in the bid dosing arm.
Further investigations into once daily dosing of enfuvirtide are warranted in larger and different patient populations (such as fully virally suppressed enfuvirtide-experienced patients) in order to fully establish the potential of qd dosing.
AUTHOR CONCLUSION
The antiviral efficacy, safety and tolerability of 180 mg enfuvirtide qd were similar to that of 90 mg enfuvirtide bid over 48 weeks of treatment.
Treatment outcomes in both arms of this exploratory pilot study appear to be similar, but due to the small number of patients in the study no inferences on the non-inferiority of qd vs. bid enfuvirtide dosing can be made.
Despite better adherence in the qd arm there was a trend favouring the bid arm for < 50 copies/mL.
Study design and entry criteria
T20-401 was a pilot Phase II, open-label, randomized, controlled study comparing the antiviral efficacy and safety of enfuvirtide 180 mg qd (two injections of 90 mg administered as close in time to each other as possible, in different injection sites) vs. enfuvirtide 90 mg bid over 48 weeks of treatment. Enfuvirtide was administered in combination with an optimized background (OB) ARV regimen in both treatment arms.
Eligibility criteria for the study included that patients should be fusion inhibitor naive, on an unchanged pre-study regimen for at least 28 days with a viral load (VL) of ≥ 5000 copies/mL, and have prior experience or documented resistance to three classes of approved oral ARVs (NRTIs, NNRTIs, PIs).
The study was designed as a Phase II pilot study (n = 120) and not powered
to demonstrate non-inferiority of qd vs. bid dosing of enfuvirtide. In February 2005, as part of an ongoing programme review, the study sponsor limited accrual to n = 60 to obtain data in a more timely manner.
A planned interim analysis when patients had completed 8 weeks on study, as well as Weeks 24 and 48 efficacy and safety were reviewed by an independent Data Safety Monitoring Board.
Efficacy analyses
The primary efficacy endpoint for the study was the proportion of patients with a viral load of < 400 copies/mL at 48 weeks.
Secondary efficacy endpoints included the proportion of patients with a viral load of < 50 copies/mL, proportion of patients with change from baseline in viral load of ≥ 1 log10 and change from baseline in viral load and CD4 count.
Virological failure was defined as:
- Plasma HIV-1 RNA < 0.5 log10 decrease from baseline at or after Week 4.
This criterion must be confirmed with a second, consecutive HIV-1 RNA measurement taken ≥ 14 days apart, or on three consecutive measurements with ≥ 14 days between the first and the third measurements, at any time after Week 4.
- The baseline value is an average of the log-transformed plasma HIV-1
RNA values from the Screen 2 and the baseline visits.
Patients in the qd arm who met the criterion for virological failure were encouraged to switch to the enfuvirtide bid arm after 8 weeks of treatment
in the original arm.
Safety analyses
Safety and tolerability comparisons between the two enfuvirtide regimens at Weeks 24 and 48 of the study were a secondary endpoint, and were conducted and recorded at each clinic visit.
- Safety parameters included clinical adverse events and selected clinical laboratory test results.
- Local injection site reactions (ISRs) were assessed for an overall grade and for individual signs and symptoms. Definitions of grading scales can be found in the footnote of this poster.
Adherence to enfuvirtide was assessed at each scheduled study visit as determined using a 4-day drug recall questionnaire (up to 11 visits over 48 weeks).
RESULTS
Study population
Sixty-four patients were enrolled at 22 sites in North America and were randomized 1:1 to the 180 mg qd and 90 mg bid enfuvirtide dosing schedules.
Of these, 61 patients received at least one dose of enfuvirtide and were included in the intent-to-treat (ITT) population. All analyses were performed using the ITT population.
Baseline characteristics were similar between the two treatment groups (Table 1).
Twelve patients in the qd arm and 10 in the bid arm (overall 36%) discontinued study medication prior to Week 48. The majority of discontinuations were not due to safety reasons. There was one death in the qd arm (suicide) which was judged by the investigator to be unrelated to treatment.
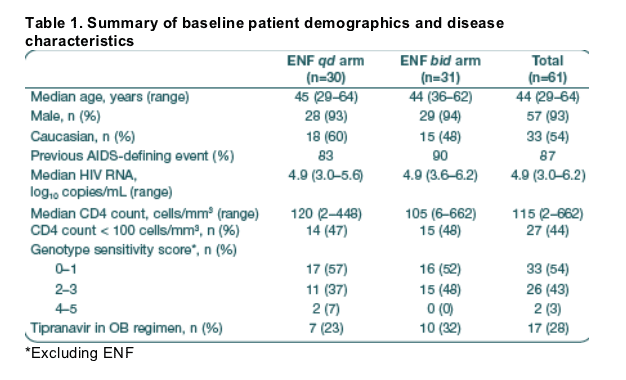
Virological response
At Week 48, the proportion of patients in both arms with a viral load of < 400 copies/mL was 23% (ITT, p = 0.97) (Figure 1).
At Week 48 using an on-treatment (OT) analysis, 64%, and 47% of patients in the qd and bid arms, respectively, achieved a viral load of < 400 copies/mL.
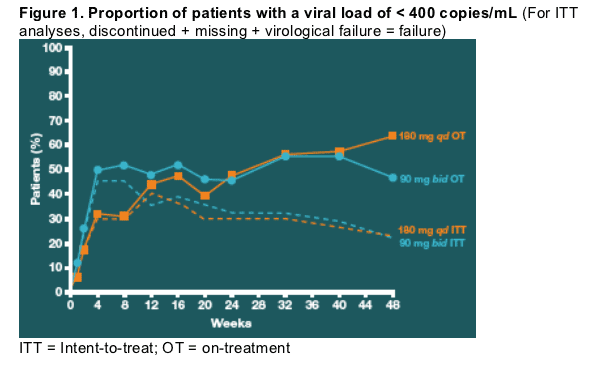
Secondary analyses are shown in Table 2. All ITT comparisons of qd vs. bid
dosing were non-significant (p > 0.05).
Table 2. Secondary efficacy endpoints at Week 48
Patients with >1 log decrease in viral load (ITT, DC+M+VF=F): 23% in qd, 32% bid; On-Treatment: 64% qd, 67% bid. Patients with viral load <50 c/ml (ITT): 13% qd, 22% bid; On-Treatment: 36% qd, 47% bid.
At Week 48 the percentage of patients with virological failure was similar in both the qd and bid arms (12/30 patients [40%] and 11/31 patients [36%], respectively).
Most virological failures occurred before Week 24 (11/12 patients [92%] in the qd arm and 9/11 patients [82%] in the bid arm), with only a small percentage of patients experiencing virological failure from Week 24 onwards.
Immunological response
Immunological responses are described in Table 2. For each of the data handling rules, the qd arm had higher CD4 responses than the bid arm, but the difference between the treatment arms was not statistically significant (p = 0.55, p = 0.55 and p = 0.18 for baseline carried forward, last observation carried forward and on-treatment analysis, respectively).
Safety analyses
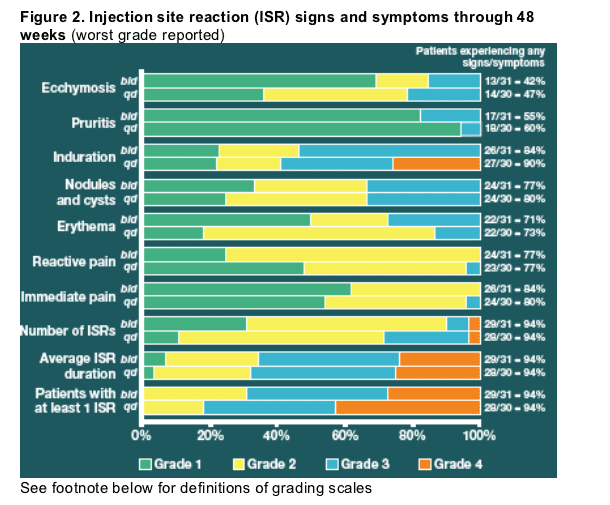
Injection site reactions
For those patients reporting at least one ISR, injection site ecchymosis, induration and erythema were moderately less severe with bid dosing (Figure 2).
One patient in the qd arm discontinued enfuvirtide due to ISRs.
Other adverse events
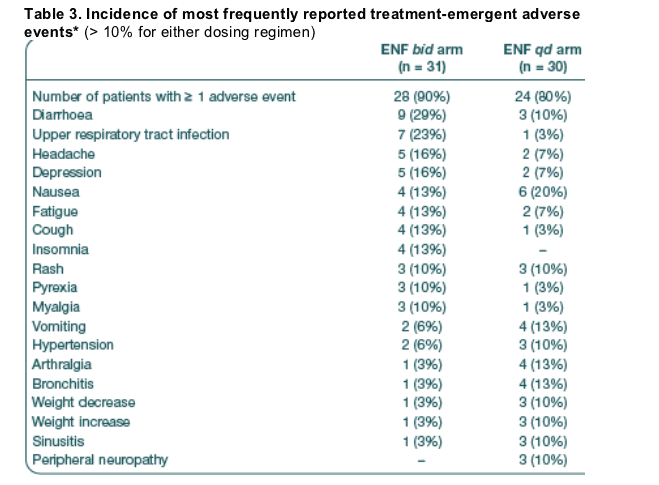
The percentage of subjects reporting at least one adverse event (excluding ISRs) was similar between the qd and bid arms (80% and 90%, respectively) (Table 3)
- Nausea was the most reported adverse event in the qd arm (20% compared
with 13% bid) followed by vomiting, arthralgia and bronchitis (all reported by 13% of qd patients).
- In the bid arm the most commonly reported adverse events were diarrhoea, upper respiratory tract infections, headache and depression (29%, 23%, 16% and 16%, respectively, compared with 10%, 3%, 7% and 7% in the qd arm).
Adherence
At Week 48, the proportion of patients reporting ≥ 85% adherence to enfuvirtide was 93% (28/30) in the qd arm compared with 81% (25/31) in the bid arm (p = 0.14).
Further analysis at Week 48 showed that 80% of patients (24/30) in the qd arm
reported ≥ 95% adherence to enfuvirtide compared with 58% of patients (18/31) in the bid arm (p = 0.06).
In the ≥ 85% adherence subgroup, 21% vs. 28% of qd and bid patients, respectively, achieved < 400 copies/mL (p = 0.58) by Week 48. Of the patients with ≥ 95% adherence to enfuvirtide, 25% compared with 28% in the qd and
bid arms, respectively, achieved < 400 copies/mL by Week 48 (p = 0.84).
REFERENCES
1. Thompson M, et al. AIDS 2006; 20:397-404.
2. Nelson M, et al. JAIDS 1995; 40:404-12.
Grading of injection site reactions
Ecchymosis: Grade 1, ≦ 20 mm; Grade 2, > 20 mm but ≦ 30 mm; Grade 3,
> 30 mm but ≦ 50 mm; Grade 4 > 50 mm.
Pruritis: Grade 1, mild not requiring treatment; Grade 2, requiring topical treatment; Grade 3, refractory to topical treatment, OR requiring oral or parenteral treatment; Grade 4, not applicable.
Induration: Grade 1, slight; Grade 2, present but < 25 mm; Grade 3, > 25 mm but ≦ 50 mm; Grade 4 > 50 mm.
Nodules or cysts: Grade 1, ≦ 20 mm; Grade 2, > 20 mm but ≦ 30 mm; Grade 3, > 30 mm; Grade 4, if draining.
Erythema: Grade 1, present but < 25 mm; Grade 2, ≥ 25 mm but < 50 mm; Grade 3 ≥ 50 mm but < 85 mm; Grade 4, ≥ 85 mm.
Reactive pain (pain that did not begin until 1 hour after injection or persisted for longer than 1 hour following injection): Grade 1, mild tenderness; Grade 2, moderate pain with limitation of usual activities; Grade 3, severe pain requiring prescription non-topical analgesics or limiting usual activities; Grade 4, not applicable.
Immediate pain (pain experienced immediately upon or within 1 hour of injection): Grade 1, mild tenderness; Grade 2, moderate pain with limitation of usual activities; Grade 3, severe pain requiring prescription non-topical analgesics or limiting usual activities; Grade 4, not applicable.
Number of ISRs: Grade 1, 1-2; Grade 2, 3-5; Grade 3, 6-14; Grade 4, ≥ 14. Average ISR duration: Grade 1, ≦ 24 hours; Grade 2, > 24 hours but ≦ 72 hours; Grade 3, > 3 days but ≦ 7 days; Grade 4, > 7 days
|
| |
|
|
|
|
|