| |
Entecavir resistance is rare in nucleoside naive patients with hepatitis B
|
| |
| |
Richard J. Colonno *, Ronald Rose, Carl J. Baldick, Steven Levine, Kevin Pokornowski, Cheng F. Yu, Ann Walsh, Jie Fang, Mayla Hsu, Charles Mazzucco, Betsy Eggers, Sharon Zhang, Mary Plym, Kenneth Klesczewski, Daniel J. Tenney
Bristol-Myers Squibb Pharmaceutical Research Institute, Wallingford, CT
Abstract
Comprehensive monitoring of genotypic and phenotypic antiviral resistance was performed on 673 entecavir (ETV)-treated nucleoside naive hepatitis B virus (HBV) patients. ETV reduced HBV DNA levels to undetectable by PCR (<300 copies/mL, <57 IU/mL) in 91% of hepatitis B e antigen (HBeAg)-positive and -negative patients by Week 96.
Thirteen percent (n = 88) of the comparator lamivudine (LVD)-treated patients experienced a virologic rebound (>1 log increase from nadir by PCR) in the first year, with 74% of these having LVD resistance (LVDr) substitutions evident. In contrast, only 3% (n = 22) of ETV-treated patients exhibited virologic rebound by Week 96.
Three ETV rebounds were attributable to LVDr virus present at baseline, with one having a S202G ETV resistance (ETVr) substitution emerge at Week 48. None of the other rebounding patients had emerging genotypic resistance or loss of ETV susceptibility.
Genotyping all additional ETV patients with PCR-detectable HBV DNA at Weeks 48, 96, or end of dosing identified seven additional patients with LVDr substitutions, including one with simultaneous emergence of LVDr/ETVr. Generally, ETV patients with LVDr were detectable at baseline (8/10) and most subsequently achieved undetectable HBV DNA levels on ETV therapy (7/10). No other emerging substitutions identified decreased ETV susceptibility.
In conclusion, ETVr emergence in ETV-treated nucleoside naive patients over a 2-year period is rare, occurring in two patients with LVDr variants. These findings suggest that the rapid, sustained suppression of HBV replication, combined with a requirement for multiple substitutions, creates a high genetic barrier to ETVr in nucleoside naive patients.
Article Text
Nearly 400 million people worldwide have chronic hepatitis B virus (HBV) infections.[1] Patients are at risk of developing progressive liver disease including fibrosis, cirrhosis, or hepatocellular carcinoma and may require liver transplantation. Approved HBV therapies include interferon, lamivudine (LVD), adefovir-dipivoxil (ADV), and entecavir (ETV).[2] However, interferon has suboptimal long-term efficacy and is contraindicated in patients with decompensated liver disease.[3] Viral resistance emerges with both LVD and ADV,[4-6] with levels as high as 70% and 29%[7][8] after 4 and 5 years, respectively. ADV can also display dose-limiting nephrotoxicity.[9] Therefore, more effective, durable, and safer therapies should be developed.
Entecavir (Baraclude, BMS-200475) is a novel deoxyguanosine analog with potent activity against HBV in vitro, in animal models, and in chronically infected patients.[10-13] In culture, ETV shows more than 100-fold greater anti-HBV potency against wild-type (WT) virus compared with LVD or ADV,[14] likely resulting from efficient and rapid intracellular phosphorylation to the active moiety, ETV-triphosphate, and inhibition of all three synthesis activities of the viral polymerase.[15][16] In long-term studies of chronic woodchuck hepatitis virus infections, ETV showed potent and sustained suppression of viral DNA and an absence of both viral rebounds and emerging ETV resistance (ETVr).[10] ETV reduced the cccDNA viral reservoir to undetectable levels and extended the lives of treated woodchucks by preventing hepatocellular carcinoma.[17] In clinical trials, ETV is highly efficacious in treating nucleoside naive and LVD refractory patients.[12][13][18][19] However, emerging resistance is a recognized complication of antiviral therapy, especially in treating chronic infections such as HBV and HIV. Therefore, monitoring for resistance becomes important to elucidate the prevalence and mechanisms of developing resistance and to more effectively manage patient treatment options.
Early evaluations of ETVr in LVD refractory patients demonstrated a requirement for pre-existing LVD resistance (LVDr) substitutions and at least one additional substitution in the viral reverse transcriptase (RT).[14] In culture, ETV susceptibility was reduced eightfold when primary LVDr substitutions L180M and M204V were present, wheras further reductions in susceptibility (70-fold) were observed with LVDr virus containing an additional change at residues T184, S202, or M250. However, these primary ETVr substitutions have not been reported in any ETV-treated patient in the absence of LVDr substitutions, and when introduced alone into recombinant clones, have a minor impact on ETV susceptibility.[20] A secondary substitution at I169 has also been observed but alone did not impact susceptibility.[14] Substitutions at residues T184, S202, or M250[14] have been detected along with LVDr changes in 6% of LVD refractory patients before ETV treatment, indicating that LVD treatment alone can result in their selection. LVDr/ETVr HBV remains susceptible to ADV[20] and virus with ADV resistance substitutions remain susceptible to ETV.[6][21]
Here, we report the cumulative results of resistance monitoring over the first 2 years of ETV treatment in both hepatitis B e antigen (HBeAg)-positive and HBeAg-negative nucleoside naive patients.[12][13]
Abbreviations
HBV, hepatitis B Virus; LVD, lamivudine; ADV, adefovir-dipivoxil; ETV, Entecavir; WT, wild-type; ETVr, entecavir resistance; LVDr, lamivudine resistance; RT, reverse transcriptase; HBeAg, hepatis B virus e antigen; PCR, polymerase chain reaction; SNP, single nucleotide polymorphism; EC50, concentration of drug required to reduce viral replication 50%; EOD, end of dosing.
Patients and Methods
Patient and Sample Disposition.
Patients treated with ETV for up to 96 weeks in three clinical studies were monitored for emergence of resistant variants. Registrational studies AI463022[13] (HBeAg-positive) and AI463027[12] (HBeAg-negative) used daily 0.5 mg ETV or 100 mg LVD treatments in nucleoside naive patients. There were 41 patients (19 and 22 randomized to ETV and LVD arms, respectively) with documented prior LVD treatment. Outcomes assessed at Week 48 of therapy determined patient designation as Responder if HBV DNA levels were undetectable by the Chiron Quantiplex bDNA assay (limit of detection 7 X 105 copies/mL), accompanied by either HBeAg loss in study AI463022 or alanine aminotransferase normalization [<1.25 X upper limit of normal (ULN)] in study AI463027, "virologic responder" (HBV DNA undetectable by bDNA without HBeAg loss or alanine aminotransferase normalization), or "non-responder" (HBV DNA detectable by bDNA). Patients designated as "responders" at week 48 stopped therapy, whereas "virologic responders" continued with blinded therapy. The remaining patients were given the option to continue on therapy in rollover study AI463901 (Table 1). On rollover, some patients received combination therapy with 100 mg LVD and 1.0 mg ETV for several months before receiving 1.0 mg ETV monotherapy. A total of 310 patients from either study, comprising 21 "responders" (all from study AI463027), 268 of the 281 'virologic responders", 17 of the 22 "non-responders", and 4 unclassified patients, continued on ETV therapy into the second year and were monitored further for evidence of resistance emergence. Only 18 patients (6%) eligible to continue treatment did not do so and could not be monitored further for resistance.
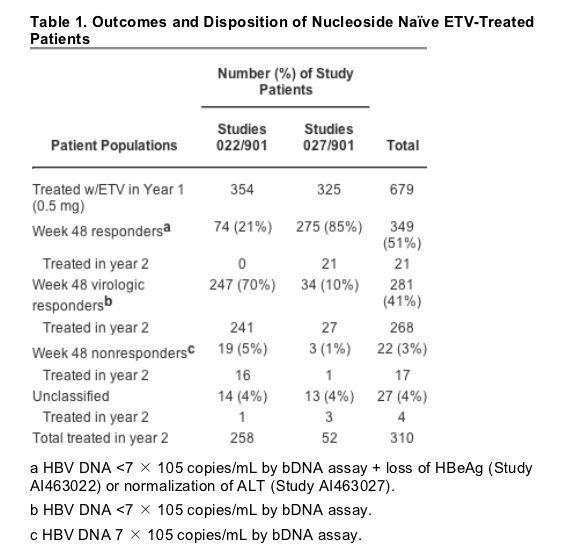
Patient Samples.
Patient serum samples were obtained from studies comparing the 48-Week safety and efficacy of 0.5 mg once daily oral (QD) ETV versus 100 mg QD LVD listed. Additionally, samples were obtained from patients who failed to reach the protocol-defined endpoints and continued therapy in rollover study AI463901 and were included in the resistance analysis only if protocol-related treatment interruption was no longer than 5 weeks (35 days). Samples designated with the capitalized "Week" were from "windowed" time points (Week 48: >42 to <58, Week 96: >90 to <102 weeks of therapy), whereas those with the lowercase "week" were from the precise time point specified.
Clinical HBV DNA Levels.
Serum HBV DNA levels were determined using the Roche COBAS Amplicor polymerase chain reaction (PCR) method (version 2.0, limit of quantification 300 copies/mL or 57 IU/mL). A virologic rebound was defined as either a confirmed (2 successive/sequential measurements while on treatment) or last on-treatment >1 log10 rise in HBV DNA levels from nadir.
HBV Polymerase Genotyping and Sequence Analysis.
HBV DNA was extracted from serum samples using commercial kits (Qiagen, Valencia, CA), and the HBV polymerase RT domain, encoding amino acids 1 to 344, was PCR amplified and sequenced directly, using primers further optimized from those described elsewhere.[14] Ultrasensitive real-time PCR-based single-nucleotide polymorphism (SNP-PCR) detection of LVDr M204 substitutions was performed essentially as described[22] using 106 copies of HBV DNA and SNP allele-specific primers that incorporate a locked nucleic acid at the 3 position.[23] Real-time PCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and SYBR Green incorporation was measured on an Applied Biosystems 9700HT. WT and LVDr mutant plasmids derived from patients were amplified and used as SNP-PCR standards.
Construction of HBV Clones and Phenotype Determination.
Methods for cloning and cell culture phenotyping of HBV isolates, using transfection of HepG2 cells with HBV expressing plasmids containing individual isolates or quasispecies populations from patient HBV RT domains were similar to those previously described.[14] Phenotyping assays were performed in the presence of a titration of antiviral compounds. Five days post-transfection, replicated, released HBV DNA was captured from the media using anti-HBV-core antibody and quantified as described.[14] Isolates from virologic rebounds were phenotyped using patient isolate quasispecies populations, whereas identified novel changes were phenotyped using individual cloned isolates. The antiviral concentration resulting in 50% inhibition of HBV was reported as the EC50.
Results
Antiviral Efficacy of ETV in Nucleoside Naive Patients.
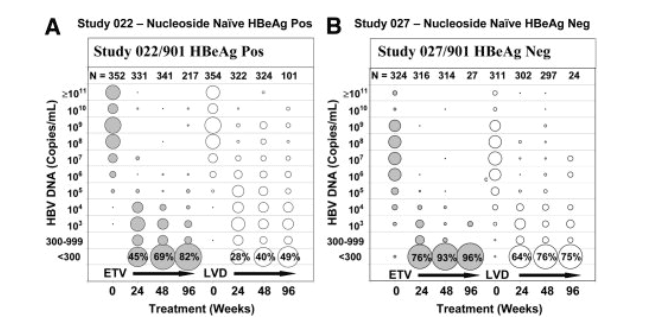
The superior in vitro potency of ETV was demonstrated in clinical studies. Rapid and substantial reductions in HBV DNA levels are exhibited in the bubble diagrams in Fig. 1, which provide an overview of the HBV DNA levels for all patients at various times on therapy. A sustained trend toward lower viral DNA levels over time on ETV therapy was observed, with PCR undetectable levels (<300 copies/mL, <57 IU/mL) achieved in 82% and 96% of HBeAg-positive and HBeAg-negative patients with on-study PCR measurements by Week 96, respectively, compared with LVD treatment reductions to undetectability in only 49% and 75% of these two patient populations, respectively, at Week 96. Cumulatively, 91% of the 664 ETV-treated HBeAg-positive and HBeAg-negative patients with PCR measurements achieved undetectable HBV DNA by Week 96. Whereas ETV continued to suppress HBV DNA levels at later times, there was evidence of a growing number of LVD-treated patients experiencing increasing HBV DNA levels at Weeks 48 and 96.
Figure 1. Nucleoside naive patient HBV DNA levels over a 2-year treatment period with ETV and LVD. Filled and open circles represent ETV- and LVD-treated patients, respectively, in HBeAg-positive studies AI463022/AI463901 (A) and HBeAg-negative studies AI463027/AI463901 (B). The size of the circles at each log10 interval is reflective of the percentage of patients (each column adds up to 100%) having the indicated viral DNA levels at that treatment time point (Week 0 = study entry, Week 24 treatment period = weeks 12-30, Week 48 treatment period = weeks 42-72, and Week 96 treatment period = weeks 90-102). The lowest circle represents patients with HBV DNA levels <300 copies/mL (<57 IU/mL), with the percentage noted in the figure. N = the number of patients with PCR measurements at each time point.
Virologic Rebounds.
Because virologic rebound (an increase of HBV DNA levels from nadir of >1 log10 by PCR) on therapy is a highly relevant clinical marker for emerging resistance, patient HBV DNA levels were monitored closely in these studies. Table 2 summarizes the number of patients who experienced a virologic rebound on ETV or LVD. Monitoring was performed through 48 Weeks for LVD- and ETV-treated patients and extended through Week 96 for ETV patients. Isolates from patients displaying virologic rebounds were analyzed for any emerging substitutions that decreased susceptibility.
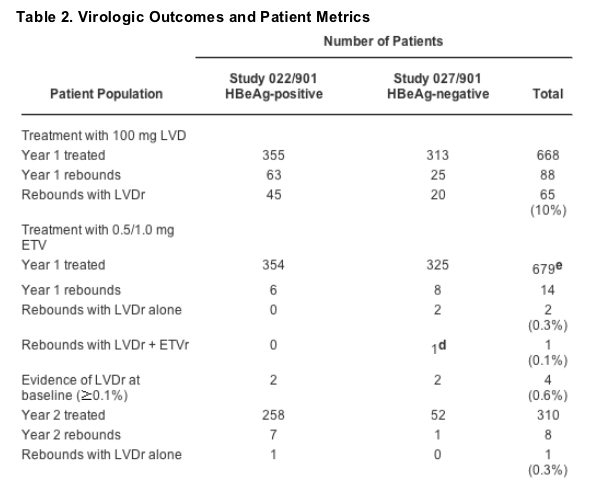

During the first year of LVD treatment, a total of 88 virologic rebounds occurred, 63 in the HBeAg-positive patients and 25 in HBeAg-negative patients. Subsequent genotypic analysis of samples from these patients found evidence of LVDr substitutions M204I/V with or without an accompanying L180M change in 65 (74%) of 88 patients (Table 2), yielding an overall virologic failure rate attributable to resistance of 10% among patients treated with LVD for 1 year. This value is somewhat lower than reported elsewhere simply because we only evaluated LVD patients experiencing virologic rebounds and did not genotype all LVD-treated patients for evidence of resistance substitutions.
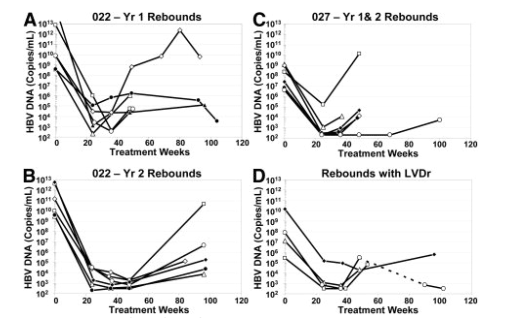
In contrast, only 14 patients (2%) experienced a virologic rebound by Week 48 on ETV therapy, with 8 (3%) additional virologic rebounds observed during the second year of treatment. Genotypic evaluation showed that 4 of the 22 patients had isolates with evidence of LVDr substitutions at the time of rebound, including one with an emerging S202G ETVr substitution (Table 3). Three of these patients had LVDr changes detectable (>0.1%) at baseline. Interestingly, none of the 22 patients had an indication of prior LVD therapy. Among the other 18 patients exhibiting a virologic rebound, only two had single emerging RT substitutions V23A or V341I. No meaningful resistance was observed for clones containing these changes in in vitro phenotypic assays (fold changes vs. WT: 1.3 for V23A and 0.6 for V341I). Thus, apart from a single patient with predominant LVDr at the time of study entry, none of the other 21 patients experiencing virologic rebound were found to harbor ETVr substitutions. Figure 2 shows the individual HBV DNA profiles for all 22 ETV-treated patients experiencing a virologic rebound through week 96 of ETV therapy. Nearly all of the rebound patients displayed significant reductions in HBV DNA levels before rebound, and only 2 of the 22 rebounds were confirmed by multiple on-treatment measurements. One patient (406) (Fig. 2A) had a subsequent on-treatment sample that did not confirm the rebound, whereas the other 19 patients had no subsequent on-treatment samples to analyze.
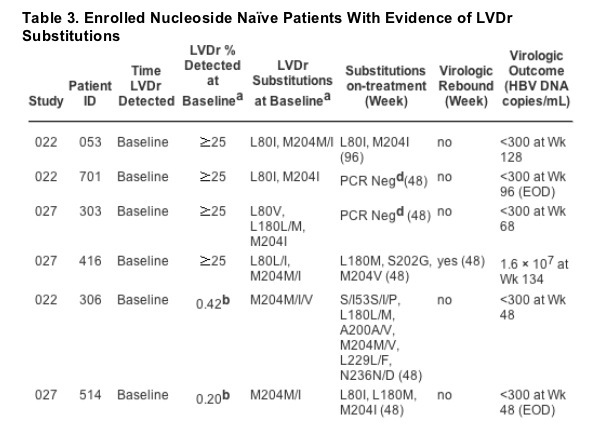

Figure 2. HBV DNA profiles of patients experiencing virologic rebounds on ETV therapy. HBV DNA levels are shown over the course of ETV treatment for patients exhibiting virologic rebounds during the first (A) and second (B) years of treatment in study AI463022/AI463901, and during the first 2 years of treatment in study AI463027/AI463901 (C), and for patients experiencing virologic rebounds due to LVDr or LVDr/ETVr viruses (D). Patient legend: Panel A (white diamond, 441; black circle, 406; black triangle, 042; white triangle, 596; white circle, 695; white square, 939); Panel B (white square, 009; white circle, 976; white diamond, 734; black diamond, 348; black circle, 346; white triangle, 671); Panel C (white square, 436; white triangle, 342; black diamond, 365; white diamond, 822; black circle, 819; white circle, 578); and Panel D (black diamond, 917; white square, 043; white triangle, 416; white circle, 713; dotted line equals treatment gap).
Population phenotypic testing of the overall susceptibility of the quasispecies from the 18 rebounding patients without evidence of LVDr substitutions showed that ETV susceptibility was virtually unchanged from baseline. Susceptibility, expressed as fold change relative to WT HBV tested in parallel (EC50/WT EC50), was approximately 1 or less. Susceptibility differences between rebound and baseline isolates were 1.8-fold or less for all but one patient (Fig. 3). This patient exhibited a 3.1-fold change due to a very low baseline susceptibility measurement, yet the rebound isolate had an EC50 that was 0.7-fold the WT clone tested in parallel.
Figure 3. Population phenotypes of HBV isolated from patients experiencing virologic rebounds on ETV therapy. Each open circle represents the population ETV susceptibility phenotype at baseline and is paired with a filled circle representing the phenotype at the time of rebound for each of the 22 patients who experienced a virologic rebound during the first 2 years of ETV therapy. Susceptibility levels are expressed relative to the laboratory WT HBV control tested in parallel. Paired phenotypes for the four LVDr patients (917, 043, 713, and 416) are as indicated. ETVr control viruses are indicated as: A, paired M204I+L180M baseline and M204I/V+L180M+T184L/I, on-treatment; B, paired M204V+L180M baseline and M204V+L180M+S202G on-treatment; and C, paired M204V+L180M baseline and M204V+L180M+T184G+S202I on-treatment.
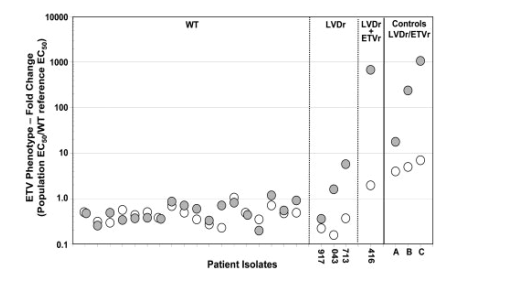
Among the four rebounding patients with LVDr substitutions, three (patients 043, 713, and 416) exhibited reduced ETV susceptibilities relative to the WT of 10-, 15- and 345-fold, respectively, as expected for viruses containing either enriched LVDr substitutions alone (patients 043 and 713, and the baseline isolates from control viruses A-C) or a combination of LVDr/ETVr changes (patient 416 and rebound isolates from control viruses A-C) (Fig. 3). The fourth, LVDr patient 917, failed to exhibit a significant change in susceptibility relative to WT virus, suggesting that phenotypic resistance was lacking because most circulating virus did not contain LVDr substitutions. This particular patient subsequently became PCR undetectable in the third year of treatment on continued ETV therapy (Table 3). In addition, none of the baseline or rebound isolates from any rebound showed significant reductions in phenotypic susceptibility to ADV (data not shown). In summary, these results indicated that in nucleoside naive patients who do not harbor LVDr HBV at the time of treatment initiation, there was no genotypic or phenotypic evidence of emerging ETVr among patients experiencing a virologic rebound while on ETV therapy.
Genotypic Surveillance for Emerging ETVr.
Signature substitutions encoding ETVr in nucleoside naive patients have not been previously identified. Therefore, our initial strategy was to evaluate randomly selected patients for emerging amino acid substitutions within the viral RT during ETV treatment by population sequencing. Ultimately, samples from all available ETV patients in studies AI463022 and AI463027 were genotyped at baseline and on-treatment if they had detectable HBV DNA (>300 copies/mL, >57 IU/mL) at Week 48, Week 96, or the End of Dosing (EOD). We were blinded to HBV DNA levels during the first year of this analysis; therefore, many of the Week 48 patient samples had HBV DNA levels that were undetectable by PCR. As a result, genotypes were successfully obtained from only 245 of the 560 patients selected for genotyping, with 131 of the 245 genotypes obtained from patients with undetectable HBV DNA levels.
This comprehensive evaluation identified four patients randomized to ETV therapy who had clear genotypic evidence of LVDr substitutions detected at baseline by standard sequencing, along with six additional patients in whom LVDr changes were first detected by sequencing at Week 48 or Week 96 (Table 3). Sequence analysis of the remaining nucleoside naive patients identified 74 amino acid residues whereas 87 emerging changes had occurred on ETV therapy (Table 4). There were no obvious patterns in the distribution of these changes, with substitutions appearing to be the result of natural genetic drift. Of the 87 changes identified, 79 appeared only once among the 245 patients sequenced, seven appeared twice, and a single substitution, S78T, was noted in three patients. None appeared to correlate with treatment responses. Most importantly, phenotypic assays using recombinant viruses containing each of the 87 changes did not result in ETV susceptibility reductions exceeding 1.8-fold the level of WT virus, indicating that these changes were unrelated to resistance development.


Characterization of Patients With LVDr Substitutions.
Because LVDr substitutions reduce ETV susceptibility eightfold and are a pre-requisite for virologic rebound because of ETVr through additional T184, S202, or M250 substitutions in LVD refractory patients, the 10 patients with these changes were extensively characterized. As discussed previously, standard sequencing of baseline samples, which detects viruses representing at least 25% of the population, identified M204I substitutions in four patients, with one (patient 416) subsequently experiencing a virologic rebound at Week 48 that coincided with the appearance of a S202G ETVr substitution (Table 3). LVDr changes became detectable by sequencing in four additional patients by Week 48, and in two patients by Week 96, with only one (patient 223) also showing evidence of an emerging S202G ETVr change. Only patient 306 reported prior LVD therapy (31 days). Four of these 10 patients experienced a virologic rebound while on ETV therapy, with two of these four patients still achieving PCR undetectable HBV DNA with continued ETV therapy (1-mg dose) (Table 3). Among the other six patients who did not experience a rebound, five also proceeded to reduce their HBV DNA levels to undetectable by PCR (Table 3) despite starting therapy with the lower 0.5-mg ETV dose recommended for nucleoside naive patients rather than the 1.0-mg dose for those with LVDr virus. The genotypes from patients with LVDr at baseline tended to show either enrichment of the LVDr change from a mixture with WT or an evolution of other LVDr substitutions over time (patients 053, 416, 306, 514, 713, 917), both likely the result of selective pressure of ETV therapy.
Because the existence of LVDr substitutions in nucleoside naive patients before LVD therapy has been reported previously,[24-30] a more sensitive PCR assay that can detect the presence of most changes at 0.1% of the population was used to further characterize the origin and timing of the emerging LVDr substitutions. Using this methodology, LVDr substitutions were detected at baseline in four of the six patients without evidence of LVDr by standard sequencing, including one (patient 306) who had received 31 days of prior LVD treatment before entering the ETV trial. Therefore, three of the four patients with LVDr substitutions at the time of rebound were attributable to pre-existing LVDr changes at baseline. The remaining two patients (043 and 223) had LVDr substitutions that were either not present or existed at levels below 0.1% at baseline. Patient 043 experienced a rebound at week 53 in the absence of ETVr substitutions, whereas patient 223 did not have a rebound but had genetically linked LVDr and ETVr substitutions appear simultaneously at week 84. Patient 223 HBV DNA levels were 2.5 X 103 and 4.6 X 103 copies/mL at Weeks 48 and 96, respectively. However, this patient experienced a rise in HBV DNA levels during the subsequent 7 weeks while off therapy and failed to respond to re-initiation of ETV therapy at week 104. It is likely that both of these patients also had pre-existing LVDr HBV variants at undetectable levels that were subsequently enriched during prolonged ETV therapy. Supporting evidence comes from the fact that the resistant variants emerged as a single isolate with either four (223) or eleven (043) genetically linked substitutions, rather than through sequential acquisition of substitutions as would be expected if these variants actually evolved de novo on therapy.
Resistance Monitoring of Patients With Detectable HBV DNA Levels at Year 2.
Additional monitoring for resistance involved genotyping all remaining patients who failed to achieve PCR undetectable HBV DNA levels by Week 96 or EOD. Apart from the 22 virologic rebounds discussed above, there were an additional 37 ETV-treated patients who had detectable HBV DNA levels by PCR at Week 96 (n = 30) or EOD before week 96 (n = 7) (Table 2) who had not been previously sequenced. Genotyping samples from these 37 patients identified five additional emerging substitutions during the second year of therapy, none of which resulted in decreased ETV susceptibility (Table 2). Interestingly, 27 of the 30 patients with detectable HBV DNA at Week 96 had HBV DNA levels >109 copies/mL (1.9 X 108 IU/mL) at study entry, which may account for the extended time needed to achieve undetectable viral DNA levels. This hypothesis is supported by the observation that 21 of the 24 patients who continued ETV therapy beyond year 2 ultimately achieved reductions in their HBV DNA to PCR undetectable levels (data not shown). Most significantly, this extended evaluation failed to produce any evidence of emerging ETVr in these patients after 2 years of therapy and confirms the high genetic barrier to resistance achieved by ETV in nucleoside naive patients without pre-existing LVDr variants.
Discussion
The ability of viruses to generate resistant variants is facilitated to a great extent by continued viral replication in the presence of an inhibitor. Thus, potent and sustained reduction of viral replication to undetectable levels should help suppress the production and emergence of resistant variants. An extensive assessment and analysis of genotypic and phenotypic resistance was performed on both HBeAg-positive and HBeAg-negative nucleoside naive patients during the first 96 weeks of treatment. The resistance profile of ETV in HBV-infected patients appears to be distinct and superior to that reported for LVD and ADV. Through 96 weeks of treatment, there is still no evidence that nucleoside naive patients who do not harbor LVDr HBV will experience a virologic rebound due to de novo ETVr. This appears to be strongly correlated with the ability of ETV to rapidly reduce circulating HBV DNA levels and to maintain this level of suppression for prolonged periods. Coupled with a requirement for multiple mutations to achieve clinically relevant resistance levels, this creates a high genetic barrier to resistance in nucleoside naive patients.
Sequencing of patient samples from studies AI463022 and AI463027 identified substitutions at the primary LVDr residue M204 in 10 patients, either at study entry or while on-treatment. However, using more sensitive detection techniques, these changes appeared to pre-exist at baseline at low levels in most, and likely all, of these patients before ETV treatment. The existence of such variants has also been reported in a small number of treatment naive patients before LVD treatment.[24-30] Interestingly, 7 of these 10 patients still exhibited reductions in their HBV DNA to PCR undetectable levels, including two of the four LVDr patients who experienced a virologic rebound, on continued ETV therapy. Only one patient (416), who had obvious LVDr substitutions at baseline by sequencing, subsequently developed a S202G ETVr substitution and proceeded to have a virologic rebound during treatment with 0.5 mg ETV.
Whether M204 LVDr substitutions are selected by ETV is of particular interest because this change could serve as a first step in developing ETVr. This change alone appears to provide partial relief to the virus from the inhibitory pressure of ETV, because LVDr HBV variants become enriched in some patients during therapy. Whether the spontaneous generation of a single LVDr change alone in an isolated virus particle would provide a pathway to ETVr is unknown; however, the odds of survival and acquiring additional substitutions in such a limited viral population would seem remote given the growth impairment observed with these viruses.[14] Selection of LVDr substitutions will continue to be monitored, but no convincing evidence has been seen that ETV can select for these substitutions in a nucleoside naive population where they do not pre-exist at some low level. Supporting this statement is the finding that only 2 of the 679 patients treated with ETV had emerging LVDr substitutions that were not detectable at baseline using an assay with a 0.1% sensitivity cutoff. Most importantly, the emerging LVDr variants that arise contained multiple substitutions that appeared simultaneously rather than sequentially, indicative of emerging viruses that pre-existed before treatment. Other novel changes leading to ETVr were not identified, since none of the other 87 genotypic changes that emerged appeared with any frequency or were associated with reduced ETV susceptibility in phenotypic assays.
The observation that virologic rebound due to ETVr likely requires several concurrent substitutions in the HBV RT, as well as overcoming the growth impairment inherit in selecting additional adaptive substitutions, is challenging in itself, but becomes even more daunting in an environment of highly suppressed viral replication achieved with ETV. In contrast to the 88 virologic rebounds observed in LVD-treated patients in year 1, fewer rebounds (n = 22) were seen on ETV therapy during the first 2 years of treatment, and none of these patients experienced subsequent biochemical failure or hepatitis flare (data not shown). Apart from the four virologic rebounds attributable to the presence of LVDr viruses and one patient with emerging LVDr/ETVr, profiling of the remaining 18 patients experiencing a virologic rebound along with the 37 ETV-treated patients (without LVDr substitutions) who had PCR-detectable HBV DNA levels by Week 96 (n = 30) or EOD (n = 7) failed to provide any evidence of emerging genotypic resistance or decreased susceptibility to ETV.
In summary, this comprehensive approach to resistance monitoring genotyped every available patient who experienced a virologic rebound or had PCR detectable HBV DNA levels at Weeks 48 or 96, or at the EOD, to find any evidence of emerging ETVr substitutions. The results failed to show any evidence of emerging ETVr in the 663 ETV-treated patients infected with HBV devoid of LVDr substitutions. However, results did show 10 patients with documented (n = 8) or circumstantial (n = 2) evidence of low levels of LVDr HBV at the time of study entry. Seven proceeded to become PCR undetectable on continued ETV therapy, and only two showed emergence of an ETVr substitution, one in the first year and one in the second year of therapy.
The combination of potent suppression of HBV replication levels coupled with the high genetic barrier is predicted to limit the emergence of resistance and enable virtually all ETV-treated patients to achieve treatment goals of reductions in HBV DNA to undetectable levels by PCR. Therefore, emergence of ETVr in nucleoside naive patients will likely require viruses containing at least three resistance substitutions either to be spontaneously selected under conditions of dramatically reduced replication levels or more likely, to arise in a very small subpopulation of patients who harbor infected cells/cccDNA that encode LVDr/ETVr viruses. In either case, these growth-impaired viruses would also need to evade functional patient immunologic responses to emerge over the course of several years of therapy.
References
1 Lee WM. Hepatitis B virus infection. N Engl J Med 1997; 337: 1733-1745. Links
2 Malik AH, Lee WM. Chronic hepatitis B virus infection: treatment strategies for the next millennium. Ann Intern Med 2000; 132: 723-731. Links
3 Fontana RJ. Management of patients with decompensated HBV cirrhosis. Semin Liver Dis 2003; 23: 89-100. Links
4 Lai CL, Dienstag J, Schiff E, Leung NW, Atkins M, Hunt C, et al. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis 2003; 36: 687-696. Links
5 Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C, et al. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology 2003; 125: 292-297. Links
6 Villeneuve JP, Durantel D, Durantel S, Westland C, Xiong S, Brosgart CL, et al. Selection of a hepatitis B virus strain resistant to adefovir in a liver transplantation patient. J Hepatol 2003; 39: 1085-1089. Links
7 Locarnini S, Qi X, Arterburn S, Snow A, Brosgart CL, Currie G, et al. Incidence and predictors of emergence of adefovir resistant HBV during four years of adefovir dipivoxil (ADV) therapy for patients with chronic hepatitis B (CHB) [Abstract]. J Hepatol 2005; 42( Suppl 2): 17. Links
8 Marcellin P, Asselah T. Resistance to adefovir: a new challenge in the treatment of chronic hepatitis B. J Hepatol 2005; 43: 920-923. Links
9 Cihlar T, Lin DC, Pritchard JB, Fuller MD, Mendel DB, Sweet DH. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol Pharmacol 1999; 56: 570-580. Links
10 Colonno RJ, Genovesi EV, Medina I, Lamb L, Durham SK, Huang ML, et al. Long-term entecavir treatment results in sustained antiviral efficacy and prolonged life span in the woodchuck model of chronic hepatitis infection. J Infect Dis 2001; 184: 1236-1245. Links
11 Innaimo SF, Seifer M, Bisacchi GS, Standring DN, Zahler R, Colonno RJ. Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob Agents Chemother 1997; 41: 1444-1448. Links
12 Lai CL, Shouval D, Lok AS, Chang TT, Cheinquer H, Goodman Z, et al. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med 2006; 354: 1011-1020. Links
13 Chang TT, Gish RG, de Man R, Gadano A, Sollano J, Chao YC, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med 2006; 354: 1001-1010. Links
14 Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, Plym M, et al. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother 2004; 48: 3498-3507. Links
15 Levine S, Hernandez D, Yamanaka G, Zhang S, Rose R, Weinheimer S, et al. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob Agents Chemother 2002; 46: 2525-2532. Links
16 Seifer M, Hamatake RK, Colonno RJ, Standring DN. In vitro inhibition of hepadnavirus polymerases by the triphosphates of BMS-200475 and lobucavir. Antimicrob Agents Chemother 1998; 42: 3200-3208. Links
17 Genovesi EV, Lamb L, Medina I, Taylor D, Seifer M, Innaimo S, et al. Efficacy of the carbocyclic 2-deoxyguanosine nucleoside BMS-200475 in the woodchuck model of hepatitis B virus infection. Antimicrob Agents Chemother 1998; 42: 3209-3217. Links
18 Chang TT, Gish RG, Hadziyannis SJ, Cianciara J, Rizzetto M, Schiff ER, et al. A dose-ranging study of the efficacy and tolerability of entecavir in Lamivudine-refractory chronic hepatitis B patients. Gastroenterology 2005; 129: 1198-1209. Links
19 Sherman M, Yurdaydin C, Sollano J, Silva M, Liaw YF, Cianciara J, et al. Entecavir for the treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 2006; 130: 2039-2049. Links
20 Colonno RJ, Rose RE, Levine SM, Pokornowski K, Plym MJ, Yu CF, et al. Entecavir (ETV) resistance is not observed in nucleoside-naive subjects and is observed infrequently by week 48 in lamivudine-refractory subjects with chronic HBV infection [Abstract]. J Hepatol 2005; 40( Suppl 2): 173. Links
21 Brunelle MN, Jacquard AC, Pichoud C, Durantel D, Carrouee-Durantel S, Villeneuve JP, et al. Susceptibility to antivirals of a human HBV strain with mutations conferring resistance to both lamivudine and adefovir. HEPATOLOGY 2005; 41: 1391-1398. Links
22 Punia P, Cane P, Teo CG, Saunders N. Quantitation of hepatitis B lamivudine resistant mutants by real-time amplification refractory mutation system PCR. J Hepatol 2004; 40: 986-992. Links
23 Latorra D, Campbell K, Wolter A, Hurley JM. Enhanced allele-specific PCR discrimination in SNP genotyping using 3 locked nucleic acid (LNA) primers. Human Mutat 2003; 22: 79-85. Links
24 Zhang X, Liu C, Gong Q, Zhang S, Zhang D, Lu Z, et al. Evolution of wild type and mutants of the YMDD motif of hepatitis B virus polymerase during lamivudine therapy. J Gastroenterol Hepatol 2003; 18: 1353-1357. Links
25 Ohishi W, Shirakawa H, Kawakami Y, Kimura S, Kamiyasu M, Tazuma S, et al. Identification of rare polymerase variants of hepatitis B virus using a two-stage PCR with peptide nucleic acid clamping. J Med Virol 2004; 72: 558-565. Links
26 Matsuda M, Suzuki F, Suzuki Y, Tsubota A, Akuta N, Hosaka T, et al. Low rate of YMDD motif mutations in polymerase gene of hepatitis B virus in chronically infected patients not treated with lamivudine. J Gastroenterol 2004; 39: 34-40. Links
27 Matsuda M, Suzuki F, Suzuki Y, Tsubota A, Akuta N, Hosaka T, et al. YMDD mutants in patients with chronic hepatitis B before treatment are not selected by lamivudine. J Med Virol 2004; 74: 361-366. Links
28 Heo J, Cho M, Kim HH, Shin YM, Jang HJ, Park HK, et al. Detection of YMDD motif mutants by oligonucleotide chips in lamivudine-untreated patients with chronic hepatitis B virus infection. J Korean Med Sci 2004; 19: 541-546. Links
29 Kobayashi S, Ide T, Sata M. Detection of YMDD motif mutations in some lamivudine-untreated asymptomatic hepatitis B virus carriers. J Hepatol 2001; 34: 584-586. Links
30 Kirishima T, Okanoue T, Daimon Y, Itoh Y, Nakamura H, Morita A, et al. Detection of YMDD mutant using a novel sensitive method in chronic liver disease type B patients before and during lamivudine treatment. J Hepatol 2002; 37: 259-265. Links
|
|
| |
| |
|
|
|