 |
|
|
| |
Resistance Workshop Review: Part 1
PI monotherapy, Boosted vs unboosted atazanavir, STIs, first-line options (NNRTIs vs boosted PI vs unboosted PI)
|
| |
| |
Written for NATAP by:
Mark Mascolini
You don't have to know how much the C-terminal domain of HIV-1 reverse transcriptase overlaps the RNase H domain to get something out of the HIV Resistance Workshop. You don't even have to know that Random Forests are turbotech computational models-not Macbeth's roving Birnam Wood-to reap useful clinical news from this annual conclave.
Sure, it helps to have an advanced degree in molecular biology or life experience with x-ray crystallography if you want to needle postdoc presenters with tough questions. But if you just want to know whether Kaletra monotherapy or borrowing your partner's efavirenz is worth the risk, this is the meeting for you.
Resistance workshoppers know they're in for a heavy dose of high science when they sign up for this get-together; some of them even seem to enjoy it. But the workshop also attracts plain old HIV docs (albeit real smart ones) who find it a useful forum to test provocative clinical findings and to pick up the latest on resistance to new antiretrovirals or the pluses and minuses of touted treatment strategies.
And this year's workshop lived up to its reputation for treating attendees to savory practical insights:
- Lopinavir/ritonavir monotherapy-either right off the bat or in an induction-maintenance scheme-caused a lot less resistance than single-dose nevirapine, for example, but a lot more than lopinavir/ritonavir backed by two nucleosides.
- Stopping an effective nonnucleoside regimen in a structured treatment interruption leaves as many as 20% of people with nonnucleoside-resistant virus-even if they stop the nonnucleoside a few days before the nukes.
- Using crystal methamphetamine before sex pushes up the risk of infection with resistant virus.
- Resistance mutations picked up from sex partners imperil chances of responding to a first-line regimen, even when those mutations are so scarce that standard resistance tests can't spot them.
This multipart review of the workshop will analyze that news-as well as trends in salvage therapy, resistance to new antiretrovirals, hepatitis virus resistance, and preventing HIV transmission-in articles posted at the NATAP Web site.
Resistance to lopinavir/ritonavir monotherapy
Until recently, resistance to first-line lopinavir/ritonavir had been rarer than syzygies. Even when an up-front lopinavir regimen failed, resistance to nucleosides in the regimen always arose first [1].
Then something happened to change that: People started taking lopinavir/ritonavir as their only antiretroviral. At the 2004 Resistance Workshop, Steven Deeks reported a case of resistance to lopinavir/ritonavir in someone who shelved his other antiretrovirals [2]. A solitary case of resistance to lopinavir/ritonavir in someone taking triple therapy has appeared in print [3].
Before these omens arose, some HIV clinicians decided to start testing lopinavir/ritonavir monotherapy, either as a first-line strategy or as maintenance therapy after shutting down viral replication with three-drug regimens. One person in each of three monotherapy studies ended up with lopinavir-resistant virus when solo lopinavir/ritonavir failed [4-6].
Teaming up with French clinicians, Abbott tested both the first-line and induction-maintenance strategies in randomized trials and reported resistance results at the workshop. (Clinical results may debut at the Toronto IAS conference.) Two people in each trial came away with protease inhibitor (PI)-resistant virus while taking unaccompanied lopinavir/ritonavir, while resistance to PIs evolved in no one taking the PIs with nucleoside backup.
The ongoing 96-week MONARK trial randomized 83 treatment-naive people to start 400/100 mg of soft-gelatin lopinavir/ritonavir twice daily and 53 to start the PIs with AZT/3TC [7]. Standard genotyping flushed out no resistant virus before treatment began. Everyone started the study with fewer than 100,000 HIV RNA copies/mL and a CD4 count of at least 100 cells/mL.
After an average 64 weeks of follow-up (range 48 to 96 weeks), poor viral control led study clinicians to order resistance tests for 21 people (25%) taking lopinavir/ritonavir monotherapy and 3 (6%) taking triple therapy. Eighteen of the 21 genotyped monotherapy patients had reached a load below 50 copies/mL then endured a rebound.
Standard genotyping found PI resistance mutations in 2 monotherapy patients and in no one taking triple therapy. The L10F and V82A mutations arose in one person, and M46I in the other. In both cases phenotyping indicated that the mutant virus remained susceptible to lopinavir. One of the 3 genotyped triple-therapy patients had resistance to 3TC, which Abbott attributed to poor adherence.
The induction-maintenance trial randomized 104 antiretroviral-naive people to start standard-dose lopinavir/ritonavir plus AZT/3TC and 51 to start efavirenz plus AZT/3TC [8]. Study participants had to have a viral load above 1000 copies/mL, but they could have any CD4 count.
Anyone in the lopinavir group who notched three consecutive viral loads below 50 copies/mL between treatment weeks 12 and 44 could drop the AZT/3TC and continue solitary lopinavir/ritonavir, and 92 did. The 12 people who didn't switch to monotherapy left the study before recording three sub-50-copy loads.
After a median 56 weeks of follow-up (range 47 to 64 weeks) in this ongoing trial, 9 of 92 monotherapy patients (10%) saw their viral load pop back up above 500 copies/mL. Genotyping disclosed resistant virus in two of them. In one person the L10F, M46L, and V82A mutations emerged in protease and the K103N and K219Q mutations in reverse transcriptase. A few weeks later genotyping also spotted I54V in protease and L210W in reverse transcriptase, but not K103N. The mutations at positions 210 and 219 confer resistance to AZT and d4T, while K103N knocks out nevirapine and efavirenz. Virus in the second person evolved the M36I, I54V, A71V, G73S, V82A, and L90M mutations in protease and M41L, D67N, V118I, and L210W in reverse transcriptase-a hefty burden of dual-class resistance.
Both of these people had pretreatment protease changes (M36I and L63P) that may or may not be provoked by therapy, as well as substitutions in reverse transcriptase (Y181C, L210W, T215Y, K219N, and K219Q). The Abbott team speculated that both people may have had tiny clusters of key protease mutations before treatment-so tiny that standard genotyping missed them. Abbott is now rechecking pretreatment samples with a more sensitive assay.
If occult pretreatment mutations become dominant populations after monotherapy began, it would suggest a need for screening with more sensitive noncommercial assays before risking PI monotherapy. To others it might suggest-more simply-abandoning PI monotherapy. Of course more discriminating resistance assays may yet disclose mutant virus in the 7 people who endured rebounds on maintenance monotherapy or whose first-line monotherapy failed in MONARK.
So where are we now in the quest to test PI monotherapy? Abbott's official conclusion to the preliminary MONARK analysis is that "although the incidence of development of PI mutations in lopinavir/ritonavir monotherapy appears to be low, the barrier for the selection of PI resistance with lopinavir/ritonavir monotherapy may be lower than with lopinavir/ritonavir-based three-drug regimens." Abbott's Michael Norton formulated a blunter conclusion at the workshop: "If you want to remain under 50 copies, three drugs are better than one."
Lopinavir/ritonavir monotherapy is nothing like the aboriginal antiretroviral regimen, AZT monotherapy. It keeps HIV under wraps in most people, at least in the short term. But these two studies and others [9,10] show that solo lopinavir/ritonavir takes second place to standard triple therapy. People tempted to try such a second-tier regimen-and clinicians tempted to test it-must ask themselves if the benefit (avoiding easy-to-take fixed-dose nucleosides) is worth the risk (viral rebound with resistant HIV).
At the workshop John Mellors (University of Pittsburgh) allowed that the induction-maintenance tactic may merit further study, "but I would completely discard the [first-line] approach."
Boosted or unboosted atazanavir?
Boosting once-daily atazanavir with ritonavir lifts atazanavir trough concentrations well above the danger zone and ensures a sustained response in previously untreated people who take their antiretrovirals faithfully. But interest in unboosted atazanavir persists, perhaps because even 100 mg of ritonavir once daily may have toxic consequences.
At the Resistance Workshop Bristol-Myers Squibb reported 48-week results of a trial that randomized 200 untreated people or people with limited antiretroviral experience to 300/100 mg of atazanavir/ritonavir or 400 mg of atazanavir once a day, plus 3TC and extended-release d4T [11]. In a refreshingly forthright conclusion, the BMS team advised that "co-administration of ritonavir [with atazanavir] may reduce the incidence of virologic failure and if virologic failure occurs, may limit the emergence of resistance mutations."
This ongoing 96-week open-label trial signed up a relatively young group (median age 34 to 35 years) with relatively advanced HIV infection. About half of the people in each treatment arm had a CD4 count under 200 cells/mL, and about half had a viral load topping 100,000 copies/mL.
Although a 48-week intent-to-treat analysis rated unboosted atazanavir "noninferior" to the boosted combo, researchers counted 10 virologic failures in the unboosted group (10%) versus 3 (3%) in the boosted group. Six people in the unboosted group had a rebound above 400 copies/mL after initial suppression, 2 never muffled viral replication, and 2 stopped the unboosted PI because of an "insufficient viral load response." The three failures in the boosted arm all resulted from rebounds.
The BMS team managed to genotype virus from 8 of 10 people in the unboosted group and 2 of 3 in the boosted group. Atazanavir's hallmark I50L mutation, which usually renders virus more susceptible to other PIs, emerged in 3 of the 8 genotyped people taking atazanavir without ritonavir. These people also had mixed viral populations at three other protease sites, 20, 33, and 71, which may or may not foster cross-resistance to other PIs [see note 12]. The 3TC-induced M184V mutation cropped up in 7 people on unboosted atazanavir. Neither of the 2 genotyped people in the atazanavir/ritonavir arm had I50L or any other PI mutation; 1 had M184V.
Justification of unboosted atazanavir seems even more dubious after this report.
One in five has resistance after nonnucleoside STI
Emergence of nonnucleoside-resistant virus during a structured treatment interruption (STI) is not news. It happened to 20% of people taking a nonnucleoside in the Italian ISS PART trial, even though researchers had study participants continue their nucleosides a few days after stopping the nonnucleoside in an attempt to prevent exposing them to slowly dwindling nonnucleoside levels after the nucleosides get flushed from the body [13]. Perhaps it is no coincidence that precisely the same proportion who interrupted their nonnucleoside in AIDS Clinical Trials Group (ACTG) protocol A5170 wound up with nonnucleoside-resistant virus [14].
As in ISS PART, the A5170 design called for continuing nucleosides a few days after suspending efavirenz or nevirapine. The problem with this stratagem is that no one knows how long to keep giving the nukes after stopping the nonnukes. If you give them too long, the threat shifts from nonnucleoside resistance due to de facto nonnucleoside monotherapy to nucleoside resistance due to nucleoside duotherapy. How long nonnucleoside levels persist after treatment stops varies widely from person to person [15,16], and research shows that efavirenz concentrations may stay high enough to provoke resistance as long as 3 weeks after people stopping taking that nonnuke [16].
This virologic substudy of A5170 focused on 33 people who suspended an efavirenz regimen and 21 who took a nevirapine combo holiday. Forty nine people took their nonnucleoside only with nucleosides, while five were also taking PIs. Everyone had a nadir (lowest-ever) CD4 count above 250 cells/mL (median 458 cells/mL), a count above 350 cells/mL when they entered the study (median 873 cells/mL), and a viral load below 400 copies/mL (43, or 80%, had fewer than 50 copies/mL). While 22 people (41%) were taking their first regimen, 32 (59%) had tried several combinations when they signed up for A5170. All 54 people in the substudy had a viral rebound above 5000 copies/mL while off therapy.
Standard genotyping spotted the K103N mutation in 5 people, all within 16 weeks of stopping their regimen. Among rebounders with no detectable mutations, more discerning allele-specific PCR assays saw K103N in 4 people, Y181C in another, and both mutations in 1 more person. All told, these assays turned up resistant virus in 11 of 54 people (20%) with a rebound after stopping suppressive nonnucleoside therapy.
Of the 5 people with K103N on standard genotyping, 4 stayed off therapy for 36 to 48 weeks, and K103N remained detectable in 3 of those 4 throughout the drug holiday. This finding underscores the persistence of nonnucleoside-induced mutations long after withdrawal of the drug.
An oligonucleotide ligation assay saw archived nonnucleoside mutations in peripheral blood cells of 6 people before the study began. But only 3 of those 6 had mutations in plasma during their drug-holiday rebound, and archived mutations did not predict resistance during rebound in a multivariate analysis. The only factor that did predict rebound resistance was a viral load between 50 and 400 copies/mL at study entry, which raised the risk of resistance 3.8 times (95% confidence interval 0.7 to 19.2, P = 0.07).
Of the 23 people who restarted treatment during the observation period, 13 started with the same regimen they stopped. Five of those 13 had virologic failures. Two people with subsequent failures returned to the same regimen even though standard genotyping saw K103N in their plasma, while 3 with later failures had no evidence of resistance mutations during their drug break.
Brad Hare (University of California, San Francisco) recommended avoiding STIs from nonnucleosides in people with a viral load between 50 and 400 copies/mL. But that proscription might extend to everyone taking a nonnuke regimen since some people with a load below 50 copies/mL when they stopped their antiretrovirals ended up with resistance to nonnucleosides.
Start with a nonnucleoside or a PI?
The merits and demerits of first-line therapy with a nonnucleoside versus a PI have provoked debate since nonnukes first challenged protease drugs as up-front options. Finally, a large randomized trial pitting PIs against nonnucleosides-appropriately named FIRST-has reached fruition. Michael Kozal (Yale University, New Haven, Connecticut) unveiled at least the resistance results at the workshop, and full response data will highlight the International AIDS Conference in August. The workshop also featured a retrospective Swiss HIV Cohort Study comparing first-off PIs and nonnucleosides.
Both FIRST and the Swiss study reached one similar conclusion: If resistance is your primary concern, base your primary regimen on PIs. But that statement oversimplifies results of both studies.
FIRST, a study run by the CPCRA trials group in the US, randomized treatment-naive people to a nucleoside regimen incorporating a PI, a nonnucleoside, or a PI plus a nonnucleoside [17]. Because the trial began over a half-decade ago, only about 25% of those taking a PI used a ritonavir boost. The resistance workshop analysis compared regimens for resistance rates at virologic failure (defined as a load above 1000 copies/mL at least 4 months after starting therapy) and reckoned how resistance affected progression to a new AIDS diagnosis or death.
But Kozal did let the clinical endpoint cat out of the bag in introducing his poster. Triple-class therapy did nothing to improve on a three-drug PI or nonnucleoside medley. For a composite endpoint including CD4-cell decline, new AIDS diagnoses, and death, the FIRST team saw no difference between PI and NNRTI combos. Virologic outcomes looked better with a nonnucleoside than with a PI, but remember, three quarters of these people took unboosted PIs-considered substandard therapy today. And the bald virologic result tells only half the tale.
Kozal analyzed outcomes in 1360 people randomized to one of the three regimens and tracked for a median of 5 years (range 44 to 78 months). The three treatment arms were well matched demographically and in pretreatment variables like viral load and CD4 count. Of 866 people with virologic failure, 266 (30.7%) had started a nonnucleoside, 328 (37.9%) a PI, and 272 (31.4%) a PI plus a nonnucleoside.
Rates for virologic failure with resistant virus measured 14.9 per 100 person-years with a PI, 10.8 per 100 person-years with a nonnuke, and 11.5 per 100 person-years with a triple-class regimen. Compared with a starting PI regimen, a first-line nonnucleoside combination lowered the risk of failure with resistant virus 22% (hazard ratio 0.78, 95% confidence interval 0.61 to 0.99).
In a statistical model considering the impact of resistance at virologic failure and the ability to push the viral load below 400 copies/mL before failure, four groups ran a significantly higher risk of AIDS or death than people without virologic failure:
- People with resistance and prior viral load suppression (hazard ratio 3.0, P < 0.001)
- People with resistance and no prior suppression (hazard ratio 4.6, P < 0.001)
- People without resistance and with prior suppression (hazard ratio 2.9, P < 0.001)
- People without resistance and with no prior suppression (hazard ratio 6.5, P < 0.001)
Among people with a resistance test during follow-up, rates of multiclass-resistant virus were higher with a nonnucleoside (34%) than with a PI (28%) or a triple-class combination (21%).
Next FIRST's number crunchers created five mutually exclusive groups according to type of resistance at virologic failure: 52 people with any resistance to PIs, 146 with resistance only to nonnucleosides, 108 with resistance only to nucleosides, 68 with resistance to nucleosides and nonnucleosides, and 475 with no resistance at failure. A statistical model adjusted for an earlier AIDS diagnosis, pretreatment CD4 count and viral load, age, resistance group, missing viral load levels, and ability to achieve virologic suppression before failure reached the following conclusion:
People who had PI-resistant virus at failure were no more likely to add a new AIDS diagnosis or to die than people who never had a virologic failure (Table 1). But people with any other type of resistance at failure were significantly more likely to endure progression to AIDS or to die than the people without failure.
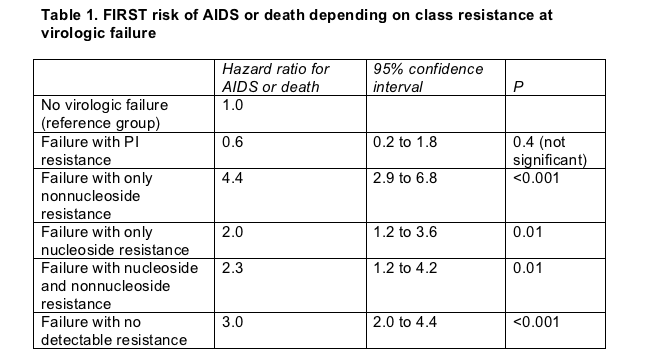
Kozal and colleagues also found that people whose regimen failed with resistance to PIs were more likely to rein in viral replication a second time than people with resistance to other antiretroviral classes at failure. "Considering the impact by resistance by class," they concluded, resistance to nonnucleosides "was the strongest predictor of disease progression."
Although predominant use of unboosted PIs surely influenced failure rates in FIRST, that anachronism should not affect progression risk with PI-resistant virus versus resistance to nonnucleosides and nucleosides. The Swiss HIV Cohort Study presented by Huldrych Gunthard (University Hospital Zurich) underlined the greater potency of boosted PIs than unboosted PIs [18]. More importantly, this illuminating analysis confirmed FIRST's finding that failure of a nonnucleoside regimen promotes more multiclass resistance than failure of a PI.
Gunthard studied 1662 treatment-naive people who started an unboosted PI, a boosted PI, or a nonnucleoside between January 1999 and December 2005. Defining virologic failure as a load at or above 500 copies/mL after 180 days of treatment, he counted 151 failures (9%). The 454 people starting a boosted PI had a lower group median CD4 count (175 cells/mL) than the 484 people starting an unboosted PI (195 cells/mL) or the 724 starting a nonnucleoside (210 cells/mL) (P = 0.03). The boosted PI group also launched their first regimen with a higher viral load (5.08 log copies/mL) than the unboosted group (4.77 log) or the nonnuke group (4.89 log) (P = 0.0001).
The Swiss team tallied 96 virologic failures (19.8%) in the unboosted PI group, compared with 42 (5.8%) in the nonnucleoside group and 13 (2.9%) in the boosted PI group. Those numbers translated into failure rates of 105 per 1000 person-years with an unboosted PI, 28.7 per 1000 person-years with a nonnucleoside, and 19.2 with a boosted PI (P < 0.0001).
Among people whose regimen failed with resistant virus, 60% in the boosted PI group had only one mutation and 10% had two or more mutations. In contrast, almost 30% in the unboosted PI group and the same proportion in the nonnuke group had two or more mutations when their regimen flopped.
Slightly fewer than 20% with a boosted PI failure had resistance to two antiretroviral classes, versus 40% who started an unboosted PI and more than 50% who started a nonnucleoside. The median number of classes affected by resistance proved significantly lower with a boosted PI (one) than with a nonnucleoside (two) or an unboosted PI (two) (P = 0.01).
Together FIRST and the Swiss HIV Cohort Study indicate that initial nonnucleoside regimens probably match boosted PIs in controlling HIV over the long term. But nonnucleoside failure exposes people to more multiclass resistance and to a bigger risk of disease progression than failure of a boosted PI.
Neither of these studies analyzed the other core variable in picking a first regimen-long-term toxicity.
Mark Mascolini writes about HIV infection (markmascolini@earthlink.net).
References and Notes
1. Kempf DJ, King MS, Bernstein B, et al. Incidence of resistance in a double-blind study comparing lopinavir/ritonavir plus stavudine and lamivudine to nelfinavir plus stavudine and lamivudine. J Infect Dis 2004;189:51-60.
2. Friend J, Parkin N, Liegler T, et al. Isolated lopinavir resistance after virological rebound of a ritonavir/lopinavir-based regimen. AIDS 2004;18:1965-1966.
3. Conradie F, Sanne I, Venter W, Eron J. Failure of lopinavir-ritonavir (Kaletra)-containing regimen in an antiretroviral-naive patient. AIDS 2004;18:1084-1085.
4. Pierone G, Mieras J, Kantor C, et al. Genotypic and phenotypic resistance observations among patients with viremia while on lopinavir/ritonavir "monotherapy." 44th ICAAC. October 30-November 2, 2004. Washington, DC. Abstract H-183.
5. Wolf E, Walter H, Eckerlein B, et al. Development of de novo PI resistance in lopinavir/ritonavir monotherapy. 3rd IAS Conference on HIV Pathogenesis and Treatment. July 24-27, 2005. Rio de Janeiro. Abstract WePe4.4C08.
6. Vanig TJ, Parkin N, Coakley EP, et al. Primary lopinavir resistance in a protease inhibitor-naive individual receiving lopinavir/ritonavir as monotherapy. 4th European HIV Drug Resistance Workshop. March 29-31, 2006. Monte Carlo. Abstract 103.
7. Norton M, Delaugere C, Batot G, et al. Drug resistance outcomes in a trial comparing
lopinavir/ritonavir (LPV/r) monotherapy to LPV/r + zidovudine/lamivudine (MONARK trial). Antivir Ther 2006;11:S84.
8. Hackett JR Jr, Holzmayer V, Marlowe N, et al. Selection of protease inhibitor (PI) resistance mutations during virological failure of lopinavir/ritonavir (LPV/r) monotherapy in an induction-maintenance study. Antivir Ther 2006;11:S85.
9. Arribas JR, Pulido F, Delgado R, et al. Lopinavir/ritonavir as single-drug therapy for maintenance of HIV-1 viral suppression. A randomized, controlled, open label, pilot, clinical trial (OK Study): 48 weeks analysis. 3rd IAS Conference on HIV Pathogenesis and Treatment. July 24-27, 2005. Rio de Janeiro. Abstract WePe12.3C05.
10. Pulido F, Arribas JR, Delgado R, et al. Risk factors for loss of virological suppression at 48 weeks in patients receiving lopinavir/ritonavir monotherapy in the OK clinical trial. 3rd IAS Conference on HIV Pathogenesis and Treatment. July 24-27, 2005. Rio de Janeiro. Abstract WePe12.3C06.
11. McGrath D, Hammond J, Frederick D, et al. Evaluation of resistance patterns in treatment-naive subjects with virological failure on atazanavir- or atazanavir/ritonavir-containing regimens. Antivir Ther 2006;11:S97.
12. The International AIDS Society (IAS)-USA lists K20M/R substitutions as secondary mutations with lopinavir, ritonavir, indinavir, and tipranavir; L33F as a primary tipranavir mutation and a secondary mutation with lopinavir or ritonavir; and A71V/T as a secondary mutation with indinavir, ritonavir, saquinavir, nelfinavir, and lopinavir. Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: 2005. Topics HIV Med 2005;13:51-57.
13. Palmisano L, Giuliano M, Bucciardini R, et al. Final results of a randomized, controlled trial of structured treatment interruptions vs continuous HAART in chronic HIV-infected subjects with persistent suppression of viral load. 13th Conference on Retroviruses and Opportunistic Infections. February 5-8, 2006. Denver. Abstract 103.
14. Hare CB, Mellors J, Krambrink A, et al. Selection of non-nucleoside reverse transcriptase inhibitor (NNRTI) resistant HIV-1 after discontinuation of a virologically suppressive regimen. Antivir Ther 2006;11:S42.
15. Ribaudo HJ, Haas DW, Tierney C, et al. Pharmacogenetics of plasma efavirenz exposure after treatment discontinuation: an Adult AIDS Clinical Trials Group study. Clin Infect Dis 2006;42:401-407.
16. Burger D, van der Heiden I, la Porte C, et al. Interpatient variability in the pharmacokinetics of the HIV non-nucleoside reverse transcriptase inhibitor efavirenz: the effect of gender, race, and CYP2B6 polymorphism. Br J Clin Pharmacol 2006;61:148-154.
17. Kozal MJ, Huppler Hellsiek K, MacArthur RD, et al. Initial virological failure with HIV drug resistance and impact of resistance on disease progression and death for patients beginning PI, NNRTI, or PI + NNRTI based strategies: the FIRST study. Antivir Ther 2006;11:S89.
18. Gunthard H, von Wyl V, Yerly S, et al. Virological failure and emergence of treatment-specific HIV-1 drug resistance patterns on first-line HAART in previously untreated patients from the Swiss HIV Cohort Study. Antivir Ther 2006;11:S82.
|
| |
|
|
|
|
|