 |
|
|
| |
Opening last week in L.A.: the era of integrase inhibition?
|
| |
| |
Reported by David Margolis, MD, University of North Carolina
CROI, Feb 2007, Los Angeles
BENCHMRK-1 and -2 studies of raltegravir:
Exciting advances are perpetually hoped for in HIV medicine, and post-Oscar anticipation peaked at CROI this year. However, the emergence of HIV integrase inhibitors as a new antiviral drug class has been expected for so long that John Mellors' proclamation that the session would be viewed as historic, made at the opening of the "Late Breaking Trials of New ARVs and Microbicides" session (Abstract 105a/bLB;
http://www.retroconference.org/2007/data/files/webpage_for_CROI.htm), seemed almost a foregone conclusion. Nevertheless, the findings of the twinned BENCHMRK-1 and -2 trials were more stirring and impressive than the Oscars that preceeded CROI.
These phase III studies of raltegravir, the baptismal name for the Merck HIV-1 integrase inhibitor previously known as MK-0518, demonstrated safety and clinical efficacy at a 16-week endpoint in patients with 3-class multidrug resistant (MDR) HIV. For those that try to keep track of generic medication designations, X-tegravir is the new moniker chosen by the FDA for HIV integrase inhibitors.
BENCHMRK-1 was implemented in Europe, Asia, the Pacific, and Peru, while BENCHMRK-2 was carried out in North and South America. This study demonstrated unprecedented success in treating this worst-case population, and with the availability of enfuvirtide, tipranavir and darunavir, and other new agents unveiled at the meeting, hopefully will usher in an era in which all patients with drug-resistant HIV who can adhere to medication can achieve full suppression of viremia and long-term clinical stability.
However, another new era may be dawning, that in which the formula 2 RTI + (PI/r or NNRTI) = ART is no longer the only simple calculation that equates to standard, initial antiretroviral therapy. While this speculation is very premature, the pursuit of the best ways to use our swelling constellation of antiretrovirals (ARVs) it will certainly occupy clinical researchers for some years to come.
To return to the data at hand and the issues of today, raltegravir is a very potent integrase inhibitor (IC95 = 33 nM) active against multidrug-resistant R5- and X4-HIV, synergistic with all ARVs in wide use, and active at week 24 in phase II studies in drug-na�ve patients (Markowitz, IAC 2006) and those with MDR HIV (Grinsztejn ICAAC 2006). Raltegravir (RGV or placebo; 2:1 weighted randomization) was given in the 400 mg. b.i.d. dose to patients with documented resistance to all three classes of ARVs and HIV RNA > 1000 copies/ml. Patients added RGV or placebo to optimized background therapy (OBT, including enfuvirtide or darunavir) and the study was evaluated at a 16-week primary endpoint. Patients receiving placebo who were failing at 16 weeks crossed over to receipt of open-label RGV. 24-week data, although incomplete, was presented and further follow-up is promised at future meetings.
500 patients were screened for BENCHMRK-1 and 512 for BENCHMRK-2, and 350 and 349 patients were randomized to study therapy, respectively. Patients were largely male (84-91%), white (55-81%), with a mean age of roughly 45 years. Mean entry CD4 cell counts ranged from 146 to 163 in the arms, and mean viral loads of 4.5-4.7 log copies/ml. More than 90% had had AIDS, with a median of 10 or more years of ARV therapy. 3-10% were hepatitis B seropositive, as were 3-20% for hepatitis C.
Resistance was advanced in this study population, with 10-30% having a genotypic or phenotypic sensitivity score of 0, meaning that none of the drug prescribed in OBT were predicted to be active by a phenotypic or genotypic resistance test, and 27-44% had a GSS or PSS of 1. In calculating this score, new receipt of enfuvirtide or darunavir added 1 point to the score. About 20% of the patients used enfuvirtide for the first time, and in the Americas study more than 25% used darunavir for the first time, while in BENCHMRK-2 nearly 50% used darunavir for the first time.
Of the 237 that received placebo, 145 have so far rolled over to open-label RGV, while of the 462 who were randomized to receipt of RGV 34 were reported to suffer virological failure by week 16. Failure was defined as a less than 1 log decline of HIV RNA and VL > 400 copies at week 16, or viral rebound of 1 log or more above nadir or rebound above 400 copies after week 16. Only 7 placebo patients have discontinued study participation (5 due to AEs) and 15 RGV patients have discontinued (9 due to AEs), a roughly equal proportion given the 2:1 randomization.
Were we living in the pre-integrase world, virologic responses in the placebo arms would seem respectable in this advanced population, with 43% achieving <400 copies/ml, and about 30% achieving < 50 copies/ml at week 16. This suggests that clinical care in the trial met current standards, newer PIs and enfuvirtide were appropriately used, and the comparable activity of RGV was not inflated by a sub-par control arm performance. However, the use of RGV doubled the response to therapy, with 79% of patients achieving < 400 copies/ml, and about 60% achieving < 50 copies/ml.
As seen in the phase II studies presented in 2006, declines of viremia appeared to be faster than seen with any previous ARV: most of the patients who would achieve < 400 copies/ml did so by week two, and most achieved <50 copies/ml by week 12 or 16. 2-log declines of RNA were generally seen in the RGV arms by week 2 or 4. CD4 count gains were also doubled in the RGV arms, with patients gaining about 75 cells by week 16.
Subgroup results delineating response in patients who were treated with enfuvirtide (ENF) and/or darunavir (DRV) for the first time were presented, giving insight into the efficacy of RGV when given (more or less) without the assistance of other potent antivirals, and when given with one or two potent drugs. 74% of patients suppressed (to <400 copies/ml) on RGV without ENF or DRV, as compared to 29% on placebo and OBT without ENF or DRV. Using either DRV or ENF with RGV yielded a 90% response, as compared to 55-63% with placebo and ENF or DRV. 87% of patients suppressed when using both ENF and DRV in OBT, and this was improved to 98% when RGV was used as well. Of note, although only about half of the patients' data could yet be reported out to 24 weeks, there did not seem to be a decline in the percentage of patients responding between weeks 16 and 24. One must hope that these responses will be durable, particularly for the patients in the study in whom RGV is the only active drug.
Of the patients with a PSS or GSS of 0, about 60% receiving RGV suppressed to <400 copies/ml at week 16, compared with 5-10% without RGV. About 70% of those with PSS or GSS of 1 responded, compared to ca. 40% without RGV, and almost 90% with a score of 2 or higher responded when given RGV, compared to 60-70% with at least 2 active drugs but without RGV.
Resistance data was presented on 41 patients with virologic failure while receiving RGV. 34 of these occurred during the first 16 weeks, and 7 occurred after 16 weeks or in placebo patients who crossed over to receive open-label RGV. Genotypes were presented from 32 of these 41 patients, as in 9 patients no consistent integrase mutations were detected. As in laboratory, pre-clinical studies, three pathways of integrase mutations were seen. N155H or Q148K or R or H were seen in most patients, and rarely mutations at Y143R or C were detected. In general, two to five "accessory" mutations were often seen with these primary mutations, thought to improve the function of the mutant enzyme.
However, one critical unknown about RGV left open to question by the data presented is the "resistance to resistance" of RGV. This is particularly of importance given the less-than-ideal results of the Gilead integrase inhibitor GS-9137 study, to be discussed later. Although a single mutation in integrase can give drug resistance, this is slow to develop in culture studies in the laboratory, as the drug-resistant integrase enzyme functions poorly. Like protease inhibitors, compensatory mutations can later develop to improve the function of drug-resistant integrase, but it is not yet clear how rapidly this occurs in clinical use. Some expert clinicians feel that integrase inhibitors should be treated like NNRTIs, to which clinical resistance can develop in days to weeks if given without sufficiently active OBT. Although this may be an appropriately conservative view to hold until more complete data is available, the experience with protease inhibitor resistance gives some hope that integrase inhibitors may prove to have some "resistance to resistance." In the BENCHMRK studies, as mentioned above, 74% of patients suppressed to <400 copies/ml on RGV without ENF or DRV. However, one would guess that most of these patients had the benefit of other active drugs in their OBT.
On the other hand, 60% of patients with GSS or PSS of 0, and therefore by definition not receiving darunavir, first-time enfuvirtide, or other drugs active as measured by resistance testing, suppressed to less than 400 copies/ml by week 16. The caveats here are that this response is only to <400 copies/ml and at 16 weeks, and that patients are likely to have "unfit" MDR virus and received multiple weakly active antivirals, which together may have augmented the RGV response.
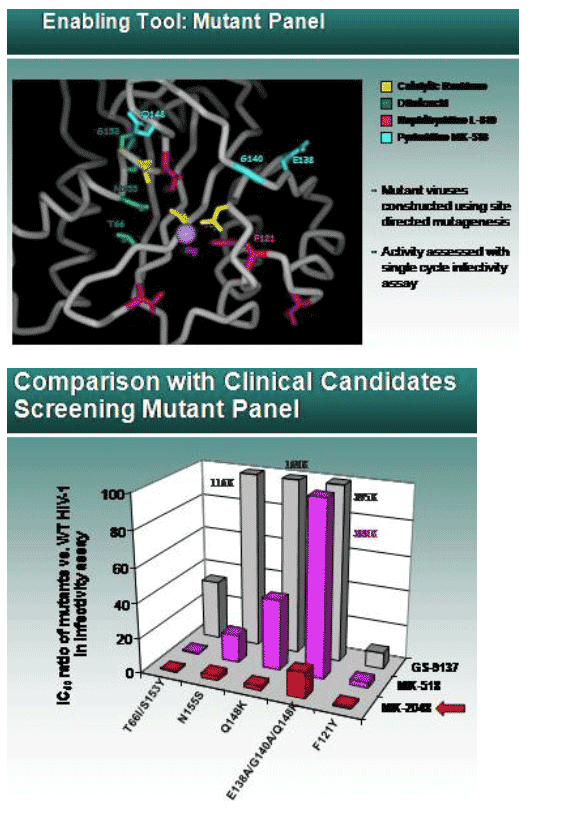
John Wai of Merck Research Laboratories presented information on resistance to integrase inhibitors in general during a separate presentation (Oral Abstracts: New ARV Agents, Resistance Mechanisms, and Clinical Resistance, Feb 27, 2007 10:00 AM; Abstract 87;
http://www.retroconference.org/2007/data/files/webpage_for_CROI.htm). Wai illustrated that when resistance mutations developed to the several generations of integrase inhibitors developed and tested by Merck over the years, the diketo acid inhibitors, the napthyridine inhibitors, and the current pyrimidine inhibitors (such as RGV), they were all positioned in adjacent areas (green, pink, and blue, respectively in Fig. 1) nearby the enzyme's catalytic active site (yellow). After the initial mutation, subsequent exposure to the drug would select for addition mutations outside of the active site area. These mutations appear to improve the drug resistant enzyme ability to function as an integrase. Using the diketo acid as a base on which to build (pharmacophore), Merck scientist subsequently added a benzyl side chain to improve antiviral activity, and a methyl side group to improve cell penetration. This candidate next-generation inhibitor (MK-2048) was found to retain activity against 3 of 4 types of drug-resistant integrase mutants. The remaining viral mutant, containing integrase mutations at E138, G140, and Q148, was somewhat resistant to MK-2048, but much more resistant to current vanguard compounds such as MK-0518 (RGV) or GS-9137 (Fig. 2).
Overall, there is no doubt that we have entered a new era in which a potent integrase inhibitor is available for use in constructing effective regimens for patients with MDR HIV. The role of RGV and other integrase inhibitors in initial therapy will await the results of a head-to-head study of RGV vs. efavirenz that will be done, and a host of other studies of novel approaches to antiviral therapy that can now be considered.
GS-9137: Gilead's integrase inhibitor in need of further development:
The next "-tegravir" is GS-9137 or elvitegravir (EGV) from Gilead Sciences. EGV is a potent antiviral with an IC50 against the integrase enzyme of 0.2 nM, and an EC90 against virus in culture of 1.2 nM. In clinical use of EGV, levels will be enhanced for once daily dosing with the coadministration of 100 mg of ritonavir. Andrew Zolopa (Abstr. 143LB) presented the results of a comparison of multiple doses of EGV to a selected PI in patients with resistance to at least 1 PI. NNRTIs were not allowed, although enfuvirtide use was.
278 patients with >1000 copies of HIV RNA/ml entered the study. 63 were randomized to the PI + optimized NRTI arm. 49% of these were prescribed darunavir and 29% prescribed tipranavir. The remaining 215 patients were divided into three dosage arms to receive 20 mg, 50 mg, and 125 mg of EGV with 100 mg of ritonavir daily. Patients were mostly male, white, in their mid-40s, with about 4.5 logs of HIV RNA/ml at entry. Mean CD4 cell counts ranged from 157 to 243/μl. Roughly half of the patients had GSS scores of 0 for the NRTIs used in OBT, with mean of 11 PI mutations, and about 20% using enfuvirtide for the first time.
At week 8 the 20 mg arm was closed by the DSMB due to a high failure rate, and due to the availability of new data showing it was safe to coadminster EGV with darunavir or tipranavir, the addition of these PIs to the EGV arms was allowed. 4 patients added a PI prior to week 16 and about 20 of the 144 patients in the remaining EGV arms added a PI.
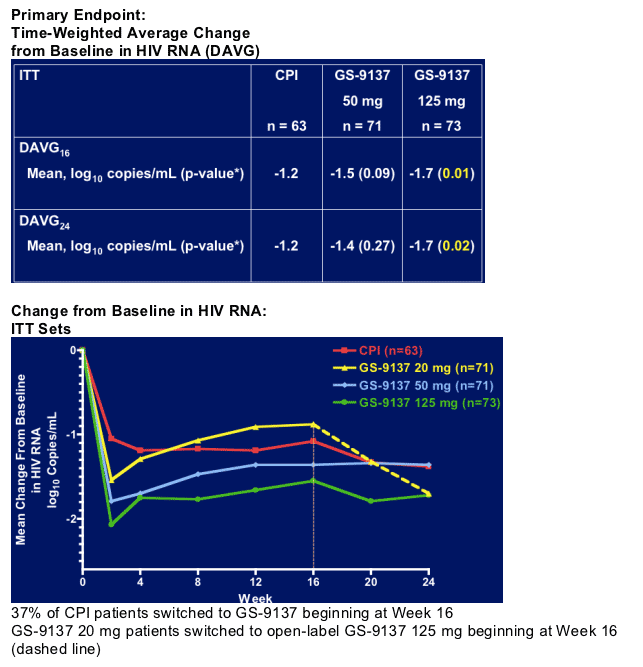
A comparison of response to EGV vs. selected PI at the week 16 time point was presented, as the addition of PIs to the EGV arm prior to the planned week 24 analysis weakened the utility of that later comparison. The mean time-weighted decline in viral load (DAVG) at week 16 and 24 was statistically superior in the 125 mg EGV arm only (Table 1, Fig. 3). The regression in the direction of baseline in all EGV arms, and the diminishing differences between the 50 and 125 mg doses, are cause for concern that dosing selected is less than ideal and/or that resistance to EGV is developing in some within this cohort of patients. Few drug-related adverse events were seen.
Resistance to EGV was discussed in a separate presentation (abstract 627).
In cell culture experiments, HIV-1 with resistance to EGC was selected by serial passage. The initial emergence of a T66I mutation in the integrase catalytic core (see Fig. 1) gave resistance to EGV. An additional mutation, R263K, located in the C-terminal DNA binding domain, was also selected. Another virus selected also encoded the T66I, and later acquired a S153Y or F121Y mutations, which conveyed phenotypic resistance to EGV. When viruses were engineered to encode the T66I, R263K, or T66I & R263K mutations they had 15.1-, 5.2-, and 98-fold resistance to EGV but were still susceptible to the napthyridine inhibitor L-870,810 (no longer in development) and RGV. However, another mutation that could be selected in integrase, E92Q, was 36-fold resistant to EGV and 7-fold resistant to RGV.
It would appear that cross-resistance within the integrase class can occur and should be guarded against. Glaxo SmithKline presented a successful phase 1 study of integrase inhibitor dosing in HIV-negative volunteers (Abstract 562). Phase II clinical studies in HIV-infected patients are said to be underway. We will see more from the integrase inhibitor class at CROI 2008.
Results of BENCHMRK-1, a Phase III Study Evaluating the Efficacy and Safety of Raltegravir (MK-0518), a Novel HIV-1 Integrase Inhibitor, in Patients with Triple-class Resistant Virus (03/05/07)
The HIV Integrase Inhibitor GS-9137 Has Potent Antiretroviral Activity in Treatment-Experienced Patients (03/01/07)
|
| |
|
|
|
|
|