| |
Valproic Acid Did Not Accelerate Decay of HIV Reservoir
|
| |
| |
Stability of the Latent Reservoir for HIV-1 in Patients Receiving Valproic Acid
The Journal of Infectious Diseases Epub Jan 30, 2007
Janet D. Siliciano,1 Jun Lai,1 Marc Callender,1 Eleanor Pitt,1 Hao Zhang,2 Joseph B. Margolick,2 Joel E. Gallant,1 Joseph Cofrancesco, Jr.,1 Richard D. Moore,1 Stephen J. Gange,3 and Robert F. Siliciano1,4
1Department of Medicine, Johns Hopkins University School of Medicine, Departments of 2Molecular Microbiology and Immunology and 3Epidemiology, Johns Hopkins Bloomberg School of Public Health, and 4Howard Hughes Medical Institute, Baltimore, Maryland
(See the editorial commentary by Schooley and Mellors following main article below)
In light of a recent report that short-term treatment with valproic acid (VA) might accelerate the decay of the latent reservoir for HIV-1 in patients receiving combination therapy and allow eventual eradication of the infection, we studied patients with prolonged suppression of viremia who were receiving combination therapy and who had also been receiving chronic VA therapy for neurological or psychiatric conditions. Latently infected cells were readily detected in all patients at levels comparable to those seen in patients receiving combination therapy alone. We conclude that the clinical use of VA has no ancillary effect on the decay of the latent reservoir.
Although highly active antiretroviral therapy (HAART) can reduce plasma HIV-1 levels to below the limit of detection in many patients, replication-competent virus persists in a stable, latent reservoir in resting CD4+ T cells [1-3]. Because of the extremely long half-life of this reservoir, it is unlikely that HIV-1 infection can be cured with ART alone [4]. Thus, there is great current interest in strategies for eliminating this reservoir. In general, these strategies involve the use of agents that will activate HIV-1 gene expression in latently infected cells so that the cells either will die from viral cytopathic effects or host cytolytic effector mechanisms or will be subject to novel therapies designed to kill infected cells [5]. HAART is continued during this process to prevent new rounds of infection by viruses released from latently infected cells that have become activated. Partial reductions in the size of the pool of latently infected cells have been reported after the use of interleukin-7 or prostratin in the SCID-hu (Thy\Liv) model of HIV-1 latency [5].
The development of strategies for efficient activation of latent HIV-1 requires a full understanding of the molecular mechanisms that restrict HIV-1 gene expression in latently infected cells. There has been considerable interest in the idea that changes in chromatin structure might limit transcription from the HIV-1 long terminal repeat (LTR). Although infected resting CD4+ T cells typically harbor HIV-1 proviruses within introns of active cellular genes [6], cis features of the LTR may allow positioning of histones in such a way that HIV-1 transcription is inhibited [7]. The binding of specific transcription factors to the LTR may allow recruitment of histone deacetylase (HDAC) 1, resulting in histone deacetylation and the repression of transcription [8, 9]. These results suggest that inhibitors of HDACs may induce HIV-1 gene expression by latently infected cells.
One currently approved drug with ancillary HDAC inhibitory activity is valproic acid (VA). It has been widely used in the treatment of seizures, migraine, and bipolar disorder [10]. The precise mechanism of action of VA in these central nervous system disorders is uncertain but may involve the modulation of neurotransmitter levels or the inhibition of voltage-dependent sodium channels. Through its activity as an HDAC inhibitor, VA can increase the expression of a variety of genes [11]. Ylisastigui et al. showed that VA could induce rescue of replication-competent HIV-1 from resting CD4+ T cells of patients receiving HAART [12]. In a recent clinical trial, 4 patients receiving HAART and the entry inhibitor enfuvirtide were given VA for 3 months, and a modest but significant decrease in the frequency of latently infected cells was noted in 3 of them [13]. This study concluded that approaches of this kind might allow the cure of HIV-1 infection.
Because of the widespread use of VA to treat common chronic neurological and psychiatric disorders, many HIV-1-infected patients have been treated simultaneously with HAART and VA for long periods of time. We therefore conducted cross-sectional and longitudinal studies of the frequency of latently infected cells in these individuals to ascertain whether the clinical use of VA had any ancillary effect on the latent reservoir for HIV-1.
Patients, materials, and methods. We studied HIV-1-infected adults who had been continuously receiving VA for at least 3 months and receiving HAART for at least 3 months with suppression of viremia to <50 copies/mL. All patients gave informed consent for donating blood.
Latently infected cells were detected by a quantitative limiting-dilution culture assay, as described elsewhere [1, 4]. Briefly, resting CD4+ T cells were purified from peripheral-blood mononuclear cells (PBMCs) by use of depletion with monoclonal antibodies and magnetic beads followed by cell sorting. This method gives resting CD4+ T cell purities of >99%. Limiting dilutions of purified resting CD4+ T cells were activated with the mitogen phyohemagglutinin and irradiated PBMCs from healthy donors. Cells were cultured in the presence of CD4+ lymphoblasts from healthy donors, to allow growth of virus released from latently infected cells. Virus growth was detected by ELISA for HIV-1 p24 antigen in the supernatant. The frequencies of infected cells were determined by the maximum likelihood method and were expressed as infectious units per million (IUPM) resting CD4+ T cells. The confidence intervals (CIs) for individual determinations were ±0.7 log10 IUPM. Random-effects regression models [14] were used to estimate the mean decrease in log10 IUPM over the follow-up time from initiation of therapy. Half-life estimates were calculated on the assumption of first-order decay kinetics.
Results.
To determine whether the clinical use of VA could affect the size of the latent reservoir for HIV-1 in patients receiving HAART, we recruited asymptomatic adult patients who had had suppression of viremia to <50 copies/mL after having received HAART for at least 3 months and who had also been receiving VA for seizures, migraine, or bipolar disorder for at least 3 months. This minimum treatment period was chosen because Lehrman et al. [13] observed a significant decay of the latent reservoir over a 3-month period in patients receiving HAART, enfuvirtide, and VA.
Patient characteristics are shown in table 1. At the time of initial sampling, the average time receiving HAART with suppression of viremia to <50 copies/mL was 26.1 months (range, 4.9-53.9 months). Plasma HIV-1 RNA levels were also measured at the time of sampling and were <50 copies/mL in all patients. At the point of initial sampling, the average time receiving VA was 21.9 months (range, 6.7-38.8 months). When initially sampled, the average length of time that the patients had been receiving both a suppressive HAART regimen and VA was 12.7 months. Thus, this group of patients had been exposed to HAART plus VA for much longer than the 3-month treatment period used in the study by Lehrman et al. It was therefore of interest to measure the frequency of latently infected cells in this cohort.
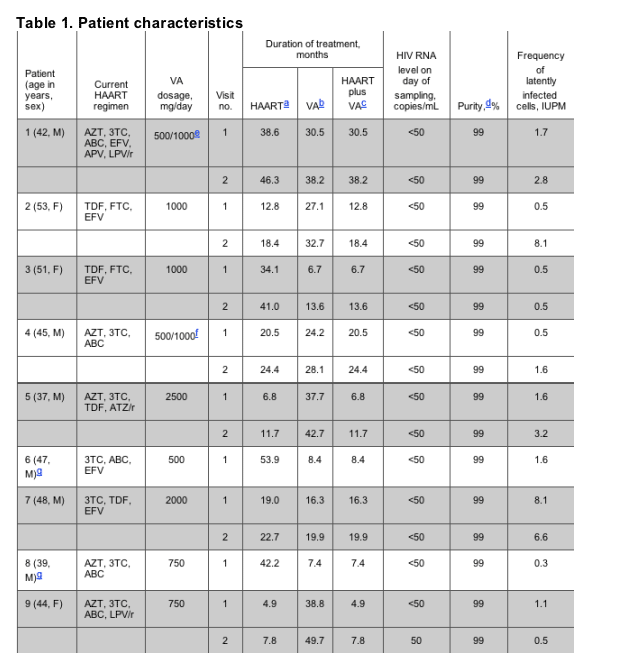
To detect latently infected cells, we used a well-characterized assay that allows measurement of the frequency of resting CD4+ T cells that are capable of releasing replication-competent virus after cellular activation [1, 4]. With this assay, latently infected cells were readily detected in all 9 patients (table 1). At the time of initial sampling, the geometric mean frequency was 1.05 IUPM, slightly higher than the mean frequency previously reported in a large cohort of patients receiving long-term suppressive HAART regimens (0.82 IUPM; [4] and studies cited therein). Thus, despite treatment with HAART plus VA for >4 times longer than the period used by Lehrman et al., levels of latently infected cells were similar to those seen in patients receiving HAART alone. It remained possible that the patients in our study had had unusually high levels of latently infected cells that had decayed to the normal range at the time of initial sampling. To exclude this possibility, repeat measurements were made in 7 of the 9 patients a mean of 5.1 months after the initial sampling. These measurements provided no evidence for decay of the latent reservoir. Frequencies actually increased in 4 patients, remained the same in 1 patient, and decreased slightly in 2 patients (table 1). The geometric mean frequency at the second sampling after a mean time receiving HAART plus VA of 19.1 months was 2.2 IUPM, approximately twice the level seen at initial sampling.
In previous studies, we have used cross-sectional and longitudinal measurements of the frequency of latently infected cells to estimate the decay rate of the latent reservoir in patients receiving long-term HAART [4]. To explore further the possibility that HAART plus VA accelerates the decay of the latent reservoir, we used the same statistical approach to estimate the decay rate of the latent reservoir in patients receiving HAART plus VA (table 2). The analysis was done using a random-effects regression model for decay with first-order kinetics. This analysis assumes that patients have different initial numbers of latently infected cells and estimates the decay rate taking into account the correlation between data from the same patient. The estimated decay rate was extremely slow, -0.00245 log10 IUPM/month (95% CI, -0.02467 to 0.01977 log10 IUPM/month). This decay rate is not statistically significant from zero (P = .7965) and is similar to the decrease seen in a large cohort of patients receiving HAART alone (-0.00681 log10 IUPM/month [4]). Use of the months receiving both HAART and VA as the time variable did not change the inferences; in fact, the estimates were then positive, with CIs that contained a zero slope. Taken together, these results suggest that the clinical use of VA in patients receiving HAART does not lead to a rapid decay in the latent reservoir.

Discussion.
We have used a well-established culture assay for latently infected cells to determine whether patients receiving HAART who are also receiving VA for neurological or psychiatric conditions experience a reduction in the size of the latent reservoir for HIV-1 in resting CD4+ T cells. Latently infected cells were readily detectable in all patients studied and did not in general decrease over time in a given patient. The estimated decay of the latent reservoir was extremely slow and was similar to that previously reported for patients receiving HAART alone.
Our results appear to differ from those of Lehrman et al. [13], who noted a decline in the size of the latent reservoir in 3 of 4 patients receiving VA for 3 months in addition to HAART and enfuvirtide. There are several potential explanations for the differences in the results of the 2 studies. First, Lehrman et al. used enfurvirtide in an effort to enhance suppression of viral replication in patients who were already responding well to HAART. Enfuvirtide was not given in our study. However, it is not clear that enfuvirtide had any additional suppressive effect on viral replication in the patients studied by Lehrman et al. In fact, 2 of the 4 patients continued to have low-level viremia after intensification of enfuvirtide. Second, the dosages of VA used in our study were not fixed. The dosages ranged from 500 to 2500 mg/day, depending on the amount needed to maintain therapeutic benefit in each patient. Lehrman et al. used dosages of 1000 to 1500 mg/day. Third, there were some differences in the assay methods used to detect latently infected cells. Finally, because latently infected cells are rare and must be quantitated by use of limiting-dilution assays that are subject to Poisson statistics, it is possible that the differences reflect statistical fluctuations in the latent-cell assays. In this type of analysis of decay curves, much greater weight can be given to points farther out on the time axis. After a mean of 19.1 months of receiving HAART plus VA and a mean of 5.1 months after initial analysis, we noted no consistent decay in the latent reservoir. Clearly, further study over prolonged treatment periods will be needed to assess the effects of VA on the latent reservoir.
Regardless of whether VA proves to be useful, efforts to decrease the size the latent reservoir through studies of the kind reported by Lehrman et al. [13] are of great importance, because cure of HIV-1 infection cannot be achieved without elimination of the latent reservoir in resting CD4+ T cells. A recent report [15] describing the rapid rebound of viremia in a patient who had discontinued HAART plus VA after >2 years of treatment highlights the need to find more-effective ways of targeting the latent reservoir. The problem is exacerbated by the exponential growth of the virus observed after treatment interruption. In principle, this allows a single residual latently infected cell to rapidly rekindle the infection. There is the additional problem that HIV-1 may persist in additional reservoirs distinct from the latent reservoir in resting CD4+ T cells.
EDITORIAL
No Cure Yet for HIV-1, But Therapeutic Research Presses On
Robert T. Schooley1 and John W. Mellors2
1Division of Infectious Diseases, Department of Medicine, University of California, San Diego, San Diego; 2Division of Infectious Diseases, University of Pittsburgh, Pittsburgh, Pennsylvania
In this issue of the Journal, Siliciano et al. [1] have used sensitive culture methods to further evaluate whether the use of valproic acid (VA), an inhibitor of histone deacetylase, might activate HIV-1 from latently infected CD4+ memory T cells, thereby reducing the size of the latent viral reservoir in persons with chronic HIV-1 infection. Although the study design has limitations that are well delineated by the authors, the work does not support findings published earlier this year by Lehrman et al. [2] suggesting that VA, combined with the fusion inhibitor enfuvirtide, accelerates the decay of the latent viral reservoir. Although VA may hold less promise than previously hoped, the goal of depleting the latent viral reservoir should continue to be vigorously pursued. A recent finding that about 80% of patients receiving suppressive antiretroviral therapy have stable, persistent viremia of 1-20 copies/mL indicates that a long-lived reservoir of virus-producing cells may also exist [3]. At a minimum, eradication of HIV-1 infection will require the complete inhibition of virus expression and the killing or permanent inactivation of long-lived latently and productively infected cells. Although daunting, the goal of curing HIV-1 infection should not be abandoned. Such is the history of antiretroviral research, which continues to make enormous strides and is worth a closer look.
It has now been 20 years since the initial demonstration that reducing viral replication in patients improves clinical outcome and 10 years since the heady early days of the highly active antiretroviral therapy era, during which there was optimism that continued viral suppression would result in elimination of virus infection in as short a period as 2-3 years [4]. As more-accurate assessments of viral reservoirs were made, however, it became clear that suppression of viral replication alone would not be sufficient to cure HIV-1 infection [5, 6]. Nevertheless, key investments in antiretroviral drug discovery and development by industry and in rigorous clinical trials conducted by government and other entities-especially the National Institute of Allergy and Infectious Diseases and the National Institute of Child Health and Human Development-have led to major improvements in the efficacy, convenience, and cost of antiretroviral therapy [7, 8]. Despite these advances, once antiretroviral therapy is initiated, most patients can expect to be receiving it for the rest of their lives. Even though existing and new classes of small molecular inhibitors of HIV replication will continue to broaden treatment alternatives and to improve prognosis, none will break the current paradigm of combination therapy for life. What, then, are the alternatives to continuous life-long therapy?
Some had hoped that it would be possible to treat people intermittently over the course of their illness, either intervening only when CD4+ T cells decreased to levels that presage an increased risk for opportunistic infection or treating patients with fixed times of therapy administration and treatment interruption. The recent recommendation by an independent data and safety monitoring board that the Strategies for Management of Anti-Retroviral Therapy study be closed on the basis of significantly greater morbidity and mortality in the treatment-interruption arm is only the most recent demonstration that intermittent therapy has substantial risks that may be prohibitive [9]. Although the results of intermittent studies have been mixed, recent trials, including several in resource-limited settings, have also cast serious doubt on the utility of this approach [10-12]. Studies of intermittent therapy will undoubtedly continue, but concerns about excess morbidity and mortality and the frequent selection of drug-resistant strains in treatment-interruption arms should provoke a careful look at trial designs and patient safety.
Whether more-prolonged treatment interruption will be possible with therapeutic vaccination remains a key area of active investigation. Several early studies have shown promise in this regard [13, 14]. Although enthusiasm has waned for therapy with interleukin (IL)-2, other contemporary approaches to immune restoration with such immune modulating agents as IL-7, IL-15, and keratinocyte growth factor hold potential and are worthy of further investigation. Clinical investigation of these agents is complicated and is best conducted by those with insights into the pathogenesis of the disease and the capacity to manage serious untoward events. As was recently shown with a ligand for CD28, engagement of the immune response can also have considerable downsides [15]. Without ongoing support by governmental and academic communities, the evaluation of potential promising biologics for the treatment of infectious disease is likely to be threatened.
Although both general and HIV-specific immune augmentation warrant ongoing research at both the basic and clinical science levels, neither of these approaches is likely to lead to elimination of the viral reservoir. Lehrman et al. are to be commended for their pilot efforts with VA [2]. In pursuing these studies, they combined a plausible hypothesis with innovative preclinical studies and intensive, pathogenesis-focused clinical investigations in what should be a model for ongoing efforts in this area. Previous studies by Prins et al. [16] and van Praag et al. [17] of IL-2- and OKT3-based approaches are also commendable, although these, too, ultimately failed to achieve the initial goal. These studies should not be seen as failures but rather as examples of hypothesis-driven clinical investigation that should lay the groundwork for future work focused on achieving a cure. There are a multitude of tools to apply to this goal, including small interfering RNA, passive cellular immune augmentation, therapeutic vaccination, stem cell biology, cellular activation with combinations of more-selective ligands-as well as combinations of these approaches.
Other than pursuing a cure, what else remains to be accomplished in therapeutic research? Although we now have >20 antiretroviral agents, transmitted and acquired drug resistance coupled with acute and chronic toxicities of many of these agents call for ongoing drug discovery and development. In light of phase 1 and 2 data, chemokine-receptor antagonists and integrase inhibitors hold considerable promise. Although the pathway to drug registration is relatively clear, experience has shown that drug approval is only the first step in learning how to best use new agents across the spectrum of the disease. Some would argue that developing this information is the responsibility of the pharmaceutical industry, but few industry-supported clinical trials can be cited that have changed clinical practice on the basis of head-to-head comparisons of specific regimens. Such studies are, nonetheless, crucial to developing an understanding of how to best use new drugs and drug combinations. Clinical trials sponsored by the National Institutes of Health have filled this gap, leading to changes in clinical practice and optimizing the use of new antiretroviral drugs. Similarly, investigation into the best use of these new agents in resource-limited settings will largely be undertaken with support from US and European governmental agencies.
With >40 million persons already infected and few bona fide prospects for the early development of an effective vaccine, continued research on optimizing therapy is essential. It would be a grave error to dismantle the therapeutic research effort before life-long therapy is a memory and an effective vaccine is in hand. At this point, it can easily be argued that ongoing investment in clinical and translational therapeutic research, particularly innovative and rigorous investigator-initiated studies, shows more promise of ending the HIV-1 epidemic than does any other approach. We must press on.
|
|
| |
| |
|
|
|