| |
Statins, High-Density Lipoprotein Cholesterol, and Regression of Coronary Atherosclerosis
|
| |
| |
JAMA. Feb 7, 2007;297:499-508.
Stephen J. Nicholls, MBBS, PhD; E. Murat Tuzcu, MD; Ilke Sipahi, MD; Adam W. Grasso, MD; Paul Schoenhagen, MD; Tingfei Hu, MS; Kathy Wolski, MPH; Tim Crowe, BS; Milind Y. Desai, MD; Stanley L. Hazen, MD, PhD; Samir R. Kapadia, MD; Steven E. Nissen, MD
Cleveland Clinic, Cleveland, Ohio.
ABSTRACT
Context Statins reduce low-density lipoprotein cholesterol (LDL-C) levels and slow progression of coronary atherosclerosis. However, no data exist describing the relationship between statin-induced changes in high-density lipoprotein cholesterol (HDL-C) and disease progression.
Objective To investigate the relationship between changes in LDL-C and HDL-C levels and atheroma burden.
Design, Setting, and Patients Post-hoc analysis combining raw data from 4 prospective randomized trials (performed in the United States, North America, Europe, and Australia between 1999 and 2005), in which 1455 patients with angiographic coronary disease underwent serial intravascular ultrasonography while receiving statin treatment for 18 months or for 24 months. Ultrasound analysis was performed in the same core laboratory for all of the studies.
Main Outcome Measure Relationship between changes in lipoprotein levels and coronary artery atheroma volume.
Results
During statin therapy, mean (SD) LDL-C levels were reduced from 124.0 (38.3) mg/dL (3.2 [0.99] mmol/L) to 87.5 (28.8) mg/dL (2.3 [0.75] mmol/L) (a 23.5% decrease; P<.001), and HDL-C levels increased from 42.5 (11.0) mg/dL (1.1 [0.28] mmol/L) to 45.1 (11.4) mg/dL (1.2 [0.29] mmol/L) (a 7.5% increase; P<.001).
The ratio of LDL-C to HDL-C was reduced from a mean (SD) of 3.0 (1.1) to 2.1 (0.9) (a 26.7% decrease; P<.001).
These changes were accompanied by a mean (SD) increase in percent atheroma volume from 39.7% (9.8%) to 40.1% (9.7%) (a 0.5% [3.9%] increase; P = .001) and a mean (SD) decrease in total atheroma volume of 2.4 (23.6) mm3 (P<.001).
In univariate analysis, mean levels and treatment-mediated changes in LDL-C, total cholesterol, non-HDL cholesterol, apolipoprotein B, and ratio of apolipoprotein B to apolipoprotein A-I were significantly correlated with the rate of atherosclerotic progression, whereas treatment-mediated changes in HDL-C were inversely correlated with atheroma progression. In multivariate analysis, mean levels of LDL-C (beta coefficient, 0.11 [95% confidence interval, 0.07-0.15]) and increases in HDL-C (beta coefficient, -0.26 [95% confidence interval, -0.41 to -0.10]) remained independent predictors of atheroma regression.
Substantial atheroma regression (>/=5% reduction in atheroma volume) was observed in patients with levels of LDL-C less than the mean (87.5 mg/dL) during treatment and percentage increases of HDL-C greater than the mean (7.5%; P<.001). No significant differences were found with regard to clinical events.
Conclusions Statin therapy is associated with regression of coronary atherosclerosis when LDL-C is substantially reduced and HDL-C is increased by more than 7.5%. These findings suggest that statin benefits are derived from both reductions in atherogenic lipoprotein levels and increases in HDL-C, although it remains to be determined whether the atherosclerotic regression associated with these changes in lipid levels will translate to meaningful reductions in clinical events and improved clinical outcomes.
INTRODUCTION
A large body of evidence supports a central role for lowering levels of low-density lipoprotein cholesterol (LDL-C) in the prevention of atherosclerotic cardiovascular disease. Randomized controlled trials have established that statin-mediated reductions in LDL-C have a favorable effect on the incidence of cardiovascular events.1-6 As a result, LDL-C lowering has become an integral component of therapeutic strategies in the prevention of cardiovascular disease.7 In particular, the use of statins has become widespread.
Recent studies have reported that high-dose statin therapy results in an incremental benefit compared with a moderate lipid-lowering strategy.8-11 Some investigators have suggested that statins also have pleiotropic properties, such as modulation of inflammation within the arterial wall, that may contribute to their beneficial effect.12-14 Accordingly, most authorities and current national guidelines emphasize reduction in LDL-C as the primary target for lipid-lowering therapy.
Monitoring atheroma progression using serial intravascular ultrasound has been used to characterize the natural history of atherosclerosis and the effect of antiatherosclerotic therapies. Serial intravascular ultrasound studies have demonstrated that reduction in LDL-C levels using statins slows the rate of atherosclerotic disease progression11, 15-16 and may induce coronary atherosclerosis regression if very low LDL-C levels are achieved.17 Collectively, these studies show a strong linear relationship between the degree of LDL-C lowering and change in atheroma volume.17
Statins have been reported to increase high-density lipoprotein cholesterol (HDL-C) levels by 5% to 15%.1-2,4-5 However, it has never been established that these small statin-induced increases in HDL-C translate into a meaningful clinical benefit. Accordingly, statins are used primarily to decrease LDL-C levels and other therapies are used to increase HDL-C levels.18-22
The current study investigated the relationship between changes in lipoprotein levels and atheroma volume in patients with coronary artery disease (CAD) who were treated with statins. This post-hoc analysis was performed by combining the results for patients enrolled in 4 prospective clinical trials that used intravascular ultrasound to determine changes in atheroma volume during treatment. The principal objective was to determine the relative contribution of statin-induced reductions in atherogenic lipoproteins and increases in HDL-C on the rate of atheroma progression.
COMMENT
Epidemiological studies demonstrate a strong relationship between levels of LDL-C and the incidence of atherosclerotic cardiovascular disease.26 Clinical trials have established that lowering LDL-C levels with statin therapy reduces the risk of cardiovascular events.1-6 Imaging studies have shown that statin-induced reductions in LDL-C slow atheroma disease progression and that achieving very low levels of LDL-C with high-intensity statin therapy may result in regression.11 More recently, reductions in levels of the inflammatory biomarker CRP in patients treated with statins have been associated with clinical benefits, including the slowing of disease progression and reduction in morbidity and mortality.12-13 Epidemiological studies also show a strong relationship between HDL-C levels and risk for cardiovascular events. While the potential interaction between low levels of HDL-C, high triglyceride levels, and cardiovascular risk has been a source of considerable debate, changes in levels of HDL-C (but not triglyceride levels) were independent predictors of changes in atheroma burden on multivariate analysis. Analysis of placebo-controlled trials reveals that the greatest benefit of statin therapy in terms of absolute risk reduction was observed in patients with the lowest baseline levels of HDL-C.1, 3-4 However, the small increases in HDL-C observed during statin therapy have never been shown to correlate with clinical outcome.
The findings from this study provide evidence that increases in HDL-C in patients treated with statins are correlated with the beneficial effect of these agents on disease progression. This is, to our knowledge, the first time that increases in HDL-C levels have been shown to be an independent predictor of a beneficial outcome with statin therapy. Furthermore, reduction of LDL-C to less than 87.5 mg/dL (2.3 mmol/L), when accompanied by an approximately 7.5% increase in HDL-C, is associated with coronary atherosclerotic regression.
These findings suggest that the increase in HDL-C that occurs during statin therapy is clinically relevant when combined with intensive lowering of LDL-C and should be considered in the selection of therapy and subsequent management of patients with CAD. The current study has implications for the development of lipoprotein-modulating therapies to treat CAD. If new therapies27 can produce increases in functional HDL-C much larger than observed in the current study, they may have the potential to substantially reduce disease burden.
These findings complement the observations that the use of nonstatin lipid-modifying therapies to increase HDL-C levels is beneficial. Despite modest increases in HDL-C levels, fibrate therapy has been demonstrated to reduce clinical event rates in some20-21 but not all28-29 trials and reduce angiographic disease progression.30 The finding that this benefit correlates with elevation of small HDL-C particles highlights the importance of modifying the functionality as well as the overall quantity of HDL-C.31 Furthermore, HDL-C elevation from the use of niacin has been reported to reduce the rate of nonfatal myocardial infarction, with a subsequent decrease in the rate of long-term mortality many years after cessation of the drug.22 When used in statin-treated patients, niacin also has been demonstrated to slow progression of carotid intimal-medial thickness18 and promote disease regression on coronary angiography.19 The current findings are, to our knowledge, the first to correlate increases in HDL-C in statin-treated patients with favorable effects on disease progression.
The finding that increases in HDL-C in statin-treated patients may play a significant role in the promotion of atheroma regression provides further impetus for the concept that HDL-C is atheroprotective. Levels of HDL-C and apoA-I inversely correlate with the incidence of cardiovascular disease in epidemiological studies.32 Increasing HDL-C levels has a beneficial effect in a wide range of atherosclerotic animal models.33-34 A recent report demonstrated that infusing reconstituted HDL-C particles containing apoA-I Milano promoted regression of coronary atherosclerosis in humans following an acute coronary syndrome.35 In the current study, the importance of modifying the number of both proatherogenic and protective lipoprotein particles is apparent. Reducing the ratio of apoB to apoA-I was the strongest lipid predictor of changes in atheroma burden in patients treated with a statin. The finding that the change in HDL-C and apoA-I, rather than achieving specific levels, is important and supports the observation that no clinical trial data currently exist to advocate a target HDL-C level for treatment in the lipid guidelines from the National Cholesterol Education Program.36
The mechanism underlying the increase in HDL-C levels observed during statin therapy is poorly understood. While it is possible that other factors influenced HDL-C levels, such as smoking, alcohol consumption, exercise, and other drugs, statin therapy does result in HDL-C elevation. Available evidence suggests that increases in HDL-C with statin therapy results from a combination of increased expression of apoA-I37 and reduced HDL remodeling as a consequence of lowering triglyceride levels.38 There is also evidence that increases in HDL-C during statin therapy may be related to decreased activity of cholesteryl ester transfer protein, likely due to depletion of levels of very low-density lipoprotein and LDL particles.39 Each of these mechanisms also influences the composition and quality of HDL-C. With increasing attention focused on the relative functionality of the HDL particle, it will be important to further elucidate the effect that statins have on both HDL-C and the functional properties of the HDL particle in relation to atheroma progression.
The relationship between lipids, inflammation, and their effect on the arterial wall has been the subject of considerable interest.40 The current analysis is limited by the lack of measurement of CRP in 2 studies. The relationship between changes in CRP and atheroma volume observed in the REVERSAL study was not found when the data were combined with that of the ACTIVATE study, in which statin therapy was not an active experimental therapy. Furthermore, the REVERSAL trial included a statin washout period, as opposed to the ACTIVATE study in which the use of statin therapy during the study reflected its use at baseline. Data on CRP were not available for the other studies. However, greater reductions in CRP were observed in patients who underwent substantial plaque regression.
The reduction in inflammation produced by statin therapy in cellular and animal studies may involve several mechanisms including a direct effect of hepatic CRP production,41 a reduction in the levels of proatherogenic and proinflammatory lipids,42 a direct anti-inflammatory effect on the arterial wall,14 and by promoting levels of HDL-C.43 Each of these effects may potentially contribute to a beneficial effect of statin therapy on the rate of plaque progression. It also remains to be determined to what degree these factors influence clinical outcome.
This study has several important limitations. This represents a post-hoc analysis that incorporates the data from patients who participated in 4 different clinical trials. The ability to combine the original data from a large number of individuals whose serial intravascular ultrasound examinations were analyzed in the same core laboratory provides a unique opportunity to investigate factors that influence the natural history of atherosclerosis. While the individual studies differed in length of time between baseline and follow-up ultrasound evaluations and required adjustment in both univariate and multivariate analysis, it remains to be determined whether atheroma progression with time is linear.
The current findings are derived from patients with CAD diagnosed at angiography performed for a clinical indication and cannot be extrapolated to the setting of primary prevention. Furthermore, the requirement for an invasive catheterization procedure for a clinical indication, variable lipid inclusion criteria, and differences between studies in terms of the statin being the active treatment or background therapy raise a number of other potential sampling biases for the current analysis. In particular, the REVERSAL study included a statin washout period and the ASTEROID study enrolled only patients who had not been treated with a statin within the preceding 12 months, whereas the CAMELOT and ACTIVATE studies enrolled patients who were taking stable doses of statins at baseline. This is likely to underscore the finding that levels of LDL-C were reduced by only 23.5% with statin therapy when the trials were combined for this analysis.
Moreover, intravascular ultrasonography provides a suboptimal characterization of plaque components. Accordingly, it remains uncertain whether simultaneous lowering of LDL-C and raising of HDL-C has an incremental beneficial effect on plaque stabilization with statins. While the results demonstrate a favorable effect of increasing levels of HDL-C, the relationship between statin-induced changes in HDL subclasses and the rate of progression of atherosclerotic plaque remains to be investigated. Similarly, the effect of differing duration of statin use prior to enrolment and potential lifestyle modifications by participants in the clinical trials remains uncertain. In addition, smoking and prior revascularization data were not collected in the ASTEROID study.
Perhaps most important, given the limited evidence correlating favorable changes in plaque progression with a reduction in clinical events, it remains to be demonstrated that simultaneous lowering of LDL-C and elevation of HDL-C translates to less clinical events. Given that all patients were treated with a statin and the number of patients in the entire cohort, it is not surprising that there was no significant relationship between groups with regard to the incidence of clinical events during the 18- to 24-month follow-up period. A large-scale clinical trial would be warranted to demonstrate that the beneficial effect on plaque progression with simultaneous lipid modulation results in fewer clinical events.
These findings may have important implications for the management of a patient with symptomatic atherosclerotic cardiovascular disease. The finding that the beneficial effect of statins on the rate of plaque progression is derived from both reducing LDL-C and increasing HDL-C complements the previous reports that the benefit of statins on both plaque progression and clinical events may be derived in part by anti-inflammatory properties. The findings also provide further evidence to support the atheroprotective properties of HDL-C and therapeutic interventions that increase its levels. Particular emphasis should be focused on the potential efficacy of emerging therapeutic strategies aimed at simultaneously lowering levels of proatherogenic lipids and increasing the level of HDL-C. Although it remains to be determined whether the atherosclerotic regression associated with changes in lipid levels observed in this study will translate to meaningful reductions in clinical events, the findings suggest that modifying the levels of both detrimental and protective lipids should be an important objective in the management of patients with established CAD.
RESULTS
Patient Population
A total of 1455 patients underwent serial assessment of atheroma burden using intravascular ultrasonography. The clinical characteristics of patients treated with a statin in each of the 4 clinical trials are shown in Table 1. The average age was 57.6 years; 73% were men; 92% were white; the average body mass index was 30; 24% were current smokers; 76% had a history of hypertension; and 19% had a history of diabetes. Lipid parameters and measures of atheroma volume at baseline and their serial change in each clinical trial are shown in Table 2.
Changes in Biochemical Parameters and Atheroma Volume
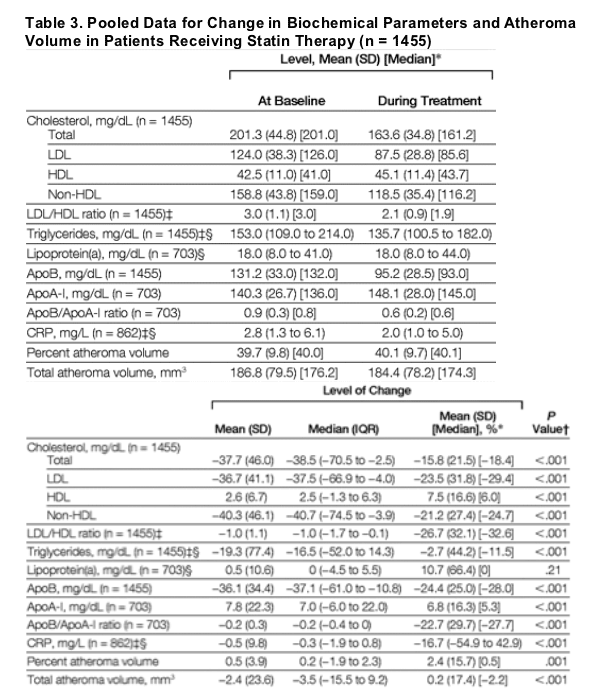
The biochemical parameters and atheroma volume at baseline and mean values during statin treatment are shown in Table 3. The mean baseline LDL-C level was 124 mg/dL (3.21 mmol/L); HDL-C level, 42.5 mg/dL (1.10 mmol/L); LDL-C/HDL-C ratio, 3.0; and median C-reactive protein (CRP), 2.8 mg/L. The mean LDL-C level during treatment was 87.5 mg/dL (2.26 mmol/L); HDL-C level, 45.1 mg/dL (1.17 mmol/L); LDL-C/HDL-C ratio, 2.1; and median CRP, 2.0 mg/L. The lipid changes translated to a mean reduction in LDL-C of 23.5% and an increase in HDL-C by 7.5%, resulting in a reduction in LDL-C/HDL-C ratio of 26.7%. Changes in all lipid parameters, except lipoprotein(a), with statin therapy were significant (P<.001 for all comparisons between baseline and completion of the study). During statin therapy, PAV increased by a mean (SD) of 0.5% (3.9%) (P = .001) and TAV decreased by 2.4 (23.6) mm3 (P<.001). As PAV expresses atheroma volume in proportion to the EEM volume, it is likely that the divergent changes in PAV and TAV reflect the effects of arterial remodeling.
Correlation Between Changes in Lipoprotein Levels and Atheroma Volume
The relationship between changes in atheroma volume and lipid levels at completion of the studies or their change is shown in Table 4. Significant correlations were found between follow-up levels of LDL-C, total cholesterol, non-HDL cholesterol, apolipoprotein B (apoB), LDL-C/HDL-C ratio, ratio of apoB to apolipoprotein A-I (apoB/apoA-I), and the changes in both PAV and TAV. These correlations remained significant when the absolute or percentage change in these lipid parameters was analyzed. Absolute and percentage changes in HDL-C level and apoA-I inversely correlated with changes in PAV and TAV.
In multivariate analysis, independent predictors of changes in PAV included baseline PAV (beta coefficient, -0.08; 95% confidence interval [CI], -0.09 to -0.06; P<.001), LDL-C level during treatment (beta coefficient, 0.02; 95% CI, 0.01-0.03; P<.001), change in HDL-C level (beta coefficient, -0.04; 95% CI, -0.06 to -0.01; P = .002), diabetes (beta coefficient, 0.60; 95% CI, 0.16-1.05; P = .008), hypertension (beta coefficient, 0.60; 95% CI, 0.15-1.06; P = .009), and age (beta coefficient, 0.02; 95% CI, 0-0.03; P = .03).
Independent predictors of changes in TAV included baseline TAV (beta coefficient, -0.06; 95% CI, -0.07 to -0.05; P<.001), LDL-C level during treatment (beta coefficient, 0.11; 95% CI, 0.07-0.15; P<.001), change in HDL-C (beta coefficient, -0.26; 95% CI, -0.41 to -0.10; P = .001), body mass index (beta coefficient, 0.33; 95% CI, 0.13-0.53; P = .001), and age (beta coefficient, 0.16; 95% CI, 0.05-0.27; P = .005).
The relationships between levels of LDL-C during treatment, percentage change of HDL-C, LDL-C/HDL-C ratio, and the absolute change in PAV are shown in the Figure. Greater percentage increases in HDL-C and lower levels of LDL-C and LDL-C/HDL-C ratio in patients during treatment with a statin resulted in atheroma regression.
The relationship between the degree of modification of both LDL-C and HDL-C is shown in Table 6. The association with the LDL-C/HDL-C ratio is shown in Table 7. Both Table 6 and Table 7 present the absolute changes in PAV and TAV and clinical events. Increases in HDL-C level greater than the mean percentage change in combination with levels of LDL-C during treatment of less than the mean percentage change were associated with the greatest degree of atheroma regression. In contrast, atheroma progression was seen in patients with percentage increases in HDL-C less than the mean and levels of LDL-C greater than the mean during treatment. No significant difference was observed between groups with regard to clinical event rates.
METHODS
Study Population
The current analysis was approved by the institutional review board of the Cleveland Clinic. All patients treated with a statin during the following intravascular ultrasound trials were included in the analysis: Reversal of Atherosclerosis With Aggressive Lipid Lowering (REVERSAL; performed at 34 sites in the United States from June 1999 to March 2003),11 Comparison of Amlodipine vs Enalapril to Limit Occurrence of Thrombosis (CAMELOT; performed at 38 sites in North America and Europe from April 1999 to April 2004),23 ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE; performed at 52 sites in the United States from December 2002 to July 2005),24 and A Study to Evaluate the Effect of Rosuvastatin on Intravascular-Ultrasound Derived Indices of Coronary Atheroma Burden (ASTEROID; performed at 53 sites in North America, Europe, and Australia from November 2002 to October 2005).17
These clinical trials evaluated serial changes in coronary atheroma burden using intravascular ultrasound in response to either high-dose statin therapy, antihypertensive therapy, or an experimental inhibitor of the enzyme acyl-coenzyme A:cholesterol acyltransferase. All patients were required to have CAD (defined as >/=20% luminal narrowing in 1 major coronary artery) on coronary angiography performed for a clinical indication. For intravascular ultrasound analyses, the target segment selected was required to have no greater than a 50% lumen narrowing for a length of at least 30 mm. The target vessel was required to have not previously undergone percutaneous coronary intervention. All participants provided written, informed consent. Race was determined by self-report by the participants.
Acquisition and Analysis of Intravascular Ultrasound Images
The details for acquisition and analysis of intravascular ultrasound images have been published.11, 17, 23-24 Coronary ultrasound was repeated at the completion of the study in the same coronary artery segment using the same experimental protocol. Analysts, unaware of both the treatment status of the patient and time point of each acquired intravascular ultrasound pullback, measured images spaced precisely 1-mm apart. The leading edges of the external elastic membrane (EEM) and lumen were traced by manual planimetry in accordance with guidelines for intravascular ultrasound of the American College of Cardiology and the European Society of Cardiology.25
Total atheroma volume (TAV) was calculated by summation of atheroma cross-sectional area (CSA) from each measured image as:

Given that the length of pullback for serial evaluations is determined by the anatomical location of the arterial side branches, there is heterogeneity in segment length between the patients. As a result, the TAV was normalized using the formula:

where n is the number of images measured in an individual pullback.
The efficacy variable of change in TAV was calculated as TAVNormal Completion of Study - TAVNormal Baseline. The percent atheroma volume (PAV) was also determined using the formula:

PAV expresses atheroma volume in proportion to the volume occupied by the entire vascular wall. The efficacy variable of change in PAV was calculated as PAVNormal Completion of Study - PAVNormal Baseline.
Biochemical Parameters
Concentrations of all biochemical parameters were determined by central laboratories (Clinical Reference Laboratory, Lenexa, Kan, for CAMELOT and Medical Research Laboratory, Highland Heights, Ky, for the other studies).
Clinical Events
Clinical event data were collected by self-report at patient visits in the ASTEROID and REVERSAL studies and by an adjudication committee in the ACTIVATE and CAMELOT studies.
Statistical Analysis
All statistical analyses were performed with SAS software version 8.2 (SAS Institute Inc, Cary, NC). For all analyses, the lipid values at all time points during treatment were averaged to provide a level during treatment. Lipid measurements were performed at patient visits (ACTIVATE, every 3 months; ASTEROID, at 3, 12, and 24 months; CAMELOT, every 6 months; REVERSAL, at 3, 6, 12, and 18 months). To account for differences in study duration (18 and 24 months), absolute changes in both PAV and TAV for individuals followed up for 24 months were multiplied by 0.75. Pearson analysis determined correlations between lipid levels during treatment and changes in lipid parameters with changes in atheroma volume. Comparisons between lipid parameters at baseline and follow-up were performed using paired t tests. Comparisons between patients who underwent substantial regression (defined as a 5% relative reduction in PAV compared with baseline) and those who did not were performed using unpaired t tests. Nonparametric tests were used when a lipid parameter was not normally distributed.
A random-effects multivariate model was performed to determine independent lipid predictors of changes in atheroma burden. This model incorporated univariate predictors of atheroma progression (baseline atheroma volume, LDL-C during treatment, change in HDL-C, age, body mass index [calculated as weight in kilograms divided by height in meters squared], hypertension, female sex, and diabetes). In addition, the random-effects model controlled for any potential influence of each trial. To test for the effect of combining the degree of modification of LDL-C and HDL-C levels on changes in atheroma volume, groups were compared by analysis of covariance after correction for baseline atheroma volume was performed. P<.05 was considered significant. Lipid parameters in patients with and without substantial atheroma regression were compared by unpaired t tests.
Financial Disclosures: Dr Nicholls reported receiving honoraria for lectures from Pfizer, AstraZeneca, and Merck Schering-Plough; consulting fees from AstraZeneca and Anthera Pharmaceuticals; and research support from LipidSciences. Dr Tuzcu reported receiving research support from Pfizer, AstraZeneca, and Sankyo Pharma; and honoraria for lectures from Pfizer and AstraZeneca. Dr Sipahi reported receiving an educational grant from Pfizer. Dr Hazen reported receiving research support and honoraria for lectures from Pfizer, Merck, and Sankyo. Dr Grasso reported owning stock in Pfizer. Dr Schoenhagen reported giving lectures for Takeda Pharmaceuticals but noted that all honoraria were directly paid to charity. Dr Desai reported receiving honoraria for lectures from Merck and Pfizer. Dr Kapadia reported receiving honoraria for lectures from Pfizer. Dr Nissen reported receiving research support from AstraZeneca, Eli Lilly, Pfizer, Takeda, Sankyo, and Sanofi-Aventis; and consulting for a number of pharmaceutical companies without financial compensation. Dr Nissen's honoraria, consulting fees, or other payments from any for-profit entity are paid directly to charity, so that neither income nor any tax deduction is received. No other authors reported financial disclosures.
|
|
| |
| |
|
|
|