| |
Once-Yearly Zoledronic Acid for Treatment of Postmenopausal Osteoporosis
|
| |
| |
NEJM May 3, 2007
Dennis M. Black, Ph.D., Pierre D. Delmas, M.D., Ph.D., Richard Eastell, M.D., Ian R. Reid, M.D., Steven Boonen, M.D., Ph.D., Jane A. Cauley, Dr.P.H., Felicia Cosman, M.D., Peter Lakatos, M.D., Ph.D., Ping Chung Leung, M.D., Zulema Man, M.D., Carlos Mautalen, M.D., Peter Mesenbrink, Ph.D., Huilin Hu, Ph.D., John Caminis, M.D., Karen Tong, B.S., Theresa Rosario-Jansen, Ph.D., Joel Krasnow, M.D., Trisha F. Hue, M.P.H., Deborah Sellmeyer, M.D., Erik Fink Eriksen, M.D., D.M.Sc., Steven R. Cummings, M.D., for the HORIZON Pivotal Fracture Trial
"....During a 3-year period, an annual infusion of 5 mg of zoledronic acid significantly reduced the risk of fracture at all key osteoporotic fracture sites, including the two primary end points, vertebral and hip fractures. The 70% reduction in the vertebral-fracture rate was greater than the 3-year reduction previously observed for oral bisphosphonates and the reductions in fracture rates associated with other antiresorptive agents .... Taken together with the sustained decrease in fracture risk, these findings support the view that the magnitude of reduction in bone remodeling associated with zoledronic acid during a 3-year period improves bone strength without adversely affecting remodeling capacity..... An increased incidence of serious atrial fibrillation was observed in the zoledronic-acid group, as compared with the placebo group..... Given the relatively poor adherence to oral bisphosphonate therapy in clinical practice, annual infusion of zoledronic acid may provide a promising approach to reducing fracture risk."
ABSTRACT
Background A single infusion of intravenous zoledronic acid decreases bone turnover and improves bone density at 12 months in postmenopausal women with osteoporosis. We assessed the effects of annual infusions of zoledronic acid on fracture risk during a 3-year period.
Methods In this double-blind, placebo-controlled trial, 3889 patients (mean age, 73 years) were randomly assigned to receive a single 15-minute infusion of zoledronic acid (5 mg) and 3876 were assigned to receive placebo at baseline, at 12 months, and at 24 months; the patients were followed until 36 months. Primary end points were new vertebral fracture (in patients not taking concomitant osteoporosis medications) and hip fracture (in all patients). Secondary end points included bone mineral density, bone turnover markers, and safety outcomes.
Results
Treatment with zoledronic acid reduced the risk of morphometric vertebral fracture by 70% during a 3-year period, as compared with placebo (3.3% in the zoledronic-acid group vs. 10.9% in the placebo group; relative risk, 0.30; 95% confidence interval [CI], 0.24 to 0.38) and reduced the risk of hip fracture by 41% (1.4% in the zoledronic-acid group vs. 2.5% in the placebo group; hazard ratio, 0.59; 95% CI, 0.42 to 0.83).
Nonvertebral fractures, clinical fractures, and clinical vertebral fractures were reduced by 25%, 33%, and 77%, respectively (P<0.001 for all comparisons).
Zoledronic acid was also associated with a significant improvement in bone mineral density and bone metabolism markers.
Adverse events, including change in renal function, were similar in the two study groups. However, serious atrial fibrillation occurred more frequently in the zoledronic acid group (in 50 vs. 20 patients, P<0.001).
Conclusions A once-yearly infusion of zoledronic acid during a 3-year period significantly reduced the risk of vertebral, hip, and other fractures. (ClinicalTrials.gov number, NCT00049829 [ClinicalTrials.gov].)
Background
Fractures are an important cause of disability among postmenopausal women, and the costs of medical care associated with osteoporosis are estimated to be more than $18 billion annually in the United States alone.1 Bisphosphonates, the most commonly used treatment for established osteoporosis, inhibit osteoclast-mediated bone resorption and reduce the risk of vertebral fracture. Two bisphosphonates, alendronate and risedronate, also have been shown to reduce nonvertebral and hip fractures in women with osteoporosis.2,3,4,5,6 However, adherence to oral treatment is problematic, and about half of patients for whom oral treatment is prescribed do not adhere to it after 1 year.7,8 Poor adherence has been shown to compromise the effectiveness of treatment against fracture and to increase the costs of medical care.9,10
A single infusion of intravenous zoledronic acid has been reported to decrease bone turnover and improve bone density for at least 12 months after infusion,11 suggesting an enduring effect. In our study, annual infusions of zoledronic acid (5 mg) for 3 years were evaluated to determine whether they reduced the risk of vertebral, hip, and other types of fracture.
Results
Baseline and Follow-up
A total of 7765 women were randomly assigned to study groups: 3889 to receive zoledronic acid and 3876 to receive placebo (Figure 1). The efficacy analysis included 7736 patients (99.6%), and the safety analysis included 7714 patients (99.3%).
The mean age of patients was 73 years, with approximately half from Europe and half from North and South America and Asia (Table 1). For measures of bone mineral density at the femoral neck, 72% of the patients had T scores below -2.5, 63% had baseline vertebral fractures, and 79% were in stratum 1. Among 1652 patients in stratum 2, the number of patients who were taking raloxifene (42%) was larger than that of patients taking any other medication. A total of 6517 patients (84%) remained in active follow-up through 3 years. A total of 6260 patients (81%) received all three infusions.
Fractures
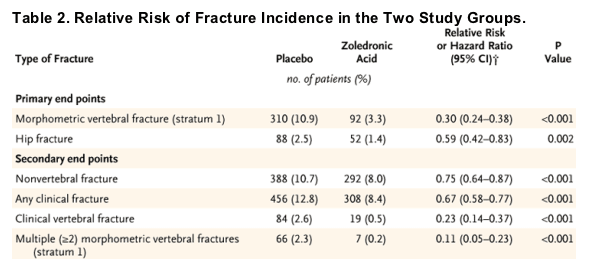
The 3-year incidence of morphometric vertebral fracture was 10.9% (involving 310 women) in the placebo group versus 3.3% (92 women) in the zoledronic-acid group, a reduction of 70% (relative risk, 0.30; 95% CI, 0.24 to 0.38) (Table 2 and Figure 2). Reductions were similar after 1 and 2 years (60% and 71%, respectively). The incidence of hip fracture was 2.5% (88 women) in the placebo group and 1.4% (52 women) in the zoledronic-acid group, a 41% reduction (hazard ratio, 0.59; 95% CI, 0.42 to 0.83) (Table 2 and Figure 2). As compared with the incidence in the placebo group, the incidence of secondary end points for fracture (including nonvertebral fractures, all clinical fractures, and clinical vertebral fractures) was significantly reduced in the zoledronic-acid group: by 25%, 33%, and 77%, respectively (P<0.001 for all comparisons). Patients in the zoledronic-acid group had significantly less height loss (-4.2 mm) than patients in the placebo group (-7.0 mm, P<0.001).
Bone Mineral Density and Biochemical Markers
In the zoledronic-acid group, bone mineral density increased significantly at the total hip (6.02%; 95% CI, 5.77 to 6.28), lumbar spine (6.71%; 95% CI, 5.69 to 7.74), and femoral neck (5.06%; 95% CI, 4.76 to 5.36), as compared with the placebo group (P<0.001 for all comparisons) (Figure 3). All three biochemical markers of bone turnover decreased significantly in patients in the zoledronic-acid group, as compared with those in the placebo group (Figure 3). At 12 months, levels of serum C-telopeptide of type I collagen, bone-specific alkaline phosphatase, and N-terminal propeptide of type I collagen were 59% (95% CI, 55 to 63), 30% (95% CI, 27 to 32), and 58% (95% CI, 55 to 60) lower, respectively, in the zoledronic-acid group (P<0.001 for all comparisons). At 6 and 12 months after each of the three infusions, the mean values were similar, with no progressive decline in marker values.
Adverse Events
The numbers of patients who died, had a serious adverse event, or discontinued follow-up because of an adverse event did not significantly differ between the study groups (Table 3), although the number of patients with adverse events was significantly higher in the zoledronic-acid group (95.5% vs. 93.9%), primarily because of a larger number of post-dose symptoms. At 9 to 11 days after infusion, 1.3% of patients in the zoledronic-acid group had an increase of more than 0.5 mg per deciliter in the serum creatinine level, as compared with 0.4% in the placebo group. However, these changes were transient; within 30 days, the levels in more than 85% of patients had returned to within 0.5 mg per deciliter of preinfusion values, and the remainder had returned to this level by the next annual follow-up. At 3 years, there was no significant difference in either serum creatinine levels or creatinine clearance between the groups. The number of patients who had any of the five most frequently reported symptoms after the first infusion was significantly higher in the zoledronic-acid group than in the placebo group, but the number with symptoms after subsequent infusions decreased substantially. Other, less common, symptoms, including chills, nausea, bone pain, and back pain, were reported more frequently in the zoledronic-acid group. These symptoms were generally rated as mild to moderate and resolved within 3 days.
The number of patients who had arrhythmia in the zoledronic-acid group (266 patients, or 6.9%) was significantly higher than that in the placebo group (203 patients, or 5.3%; P=0.003). Serious atrial fibrillation, a subcategory of all arrhythmias, was more common among patients in the zoledronic-acid group. A total of 50 patients in the zoledronic-acid group had serious atrial fibrillation (1.3%), as compared with 20 patients (0.5%) in the placebo group (P<0.001). After adjudication, the number of patients whose atrial fibrillation was reported as a serious adverse event did not change appreciably (50 in the zoledronic-acid group and 17 in the placebo group). Among the 50 patients, the events occurred more than 30 days after infusion in 47 patients. Among 559 patients who underwent electrocardiography, the prevalence of atrial fibrillation (2.1% in the zoledronic-acid group and 2.8% in the placebo group) and other electrocardiographic abnormalities did not differ significantly between the study groups. No difference was observed in the occurrence of the serious adverse events of stroke (2.3% in both study groups); the incidence of death due to stroke was 0.5% in the zoledronic-acid group and 0.3% in the placebo group (P=0.15) (Table 3).
There were no spontaneous reports of osteonecrosis of the jaw. From a search of the trial database of adverse events, which was followed by expert adjudication, two cases of potential osteonecrosis of the jaw were identified (one in the placebo group and one in the zoledronic-acid group). In both patients, delayed healing followed surgical manipulation, and both cases subsequently resolved with antibiotic therapy and debridement. A similar search for and review of osteonecrosis of the hip or knee revealed seven cases (three in the placebo group and four in the zoledronic-acid group). No adverse effect on fracture healing was observed, with three cases of nonunion (one in the placebo group and two in the zoledronic-acid group).
At 9 to 11 days after the first infusion, 49 patients in the zoledronic-acid group had a serum calcium level of less than 2.075 mmol per liter, as compared with 1 patient in the placebo group. All events were transient and asymptomatic. Patients who were treated with zoledronic acid had an absolute increase of approximately 0.69% (3.34% vs. 2.65%) in inflammatory ocular adverse events (mainly conjunctivitis) during the first 15 days after infusion.
Discussion
During a 3-year period, an annual infusion of 5 mg of zoledronic acid significantly reduced the risk of fracture at all key osteoporotic fracture sites, including the two primary end points, vertebral and hip fractures. The 70% reduction in the vertebral-fracture rate was greater than the 3-year reduction previously observed for oral bisphosphonates (40 to 50%)2,4,6,18,19,20 and the reductions in fracture rates associated with other antiresorptive agents.21,22,23 All other prospectively defined categories of fracture, including nonvertebral fractures and clinical vertebral fractures, were also significantly reduced (P<0.001 for all comparisons).
A regimen of infusions once a year appears to ensure that patients will have a full treatment effect for at least 12 months. In contrast, many patients who receive prescriptions for oral bisphosphonates stop treatment, and most appear to be taking less than 80% of their prescribed pills by 12 months.7,9,24,25,26 Adherence to a regimen of oral bisphosphonates is challenging because the drug must be taken with a full glass of water when the patient is fasting, and the patient must remain upright for at least 30 minutes after taking the medication. Since poor adherence reduces the antifracture efficacy,9 a single annual infusion of zoledronic acid might improve such efficacy in clinical practice.
The effect of zoledronic acid on biochemical markers in our study was similar to that reported for oral bisphosphonates.4,19,20,27,28,29,30 Furthermore, levels of bone remodeling after the second and third infusions were similar to levels after the first infusion. Taken together with the sustained decrease in fracture risk, these findings support the view that the magnitude of reduction in bone remodeling associated with zoledronic acid during a 3-year period improves bone strength without adversely affecting remodeling capacity.
As reported with other bisphosphonates that are administered intravenously, mild-to-moderate post-dose symptoms occurred most commonly after the first infusion. These symptoms typically resolved within 3 days after their onset and declined markedly with subsequent infusions. Treatment with antipyretic analgesics (e.g., ibuprofen and acetaminophen) appeared to mitigate these symptoms.
The study protocol included monitoring for adverse renal effects 9 to 11 days after each infusion in more than 5000 patients; only small and transient increases in serum creatinine levels were observed. At 3 years, there was no systematic deterioration in estimated creatinine clearance.
There was a small increase in the risk of inflammatory ocular adverse events within the first 15 days after infusion, as reported with other bisphosphonates.31 All such events were treated and resolved with outpatient treatment.
Most cases of osteonecrosis of the jaw have been observed in patients with cancer who were treated with frequent doses of intravenous bisphosphonates.32,33,34,35 Our study assessed the incidence of osteonecrosis of the jaw prospectively in a large number of women with osteoporosis who were receiving a bisphosphonate. There were no spontaneous reports of osteonecrosis of the jaw by patients in our study. In addition, blinded adjudication of the safety database yielded one case in the zoledronic-acid group and one in the placebo group, suggesting that the risk of osteonecrosis of the jaw in women with postmenopausal osteoporosis is very low and that this disorder may occur without bisphosphonate treatment. A once-yearly regimen with intravenous zoledronic acid does not appear to affect the frequency of this adverse event.
An increased incidence of serious atrial fibrillation was observed in the zoledronic-acid group, as compared with the placebo group. The events were uniformly distributed over time, with the vast majority of events occurring more than 30 days after infusion, by which time zoledronic acid is undetectable in the circulation. There are no studies establishing biologic mechanisms that might link bisphosphonate therapy to atrial fibrillation or arrhythmia. Alterations in serum calcium levels could be related to atrial fibrillation, but the administration of zoledronic acid had little or no effect on serum calcium levels measured 9 to 11 days after infusion. Electrocardiography that was performed before and 9 to 11 days after the third infusion in 559 patients who were not taking drugs that cause prolongation of QT intervals showed no significant difference between groups in the prevalence of arrhythmia. However, low-frequency, intermittent arrhythmia might not have been captured on the short echocardiogram in this subgroup. An increased risk of serious atrial fibrillation had not been previously associated with zoledronic acid or other bisphosphonates, although a letter in this issue of the Journal36 reports a similar, though nonsignificant, trend from the 1997 Fracture Intervention Trial of alendronate. The observed association might be due to chance but should be further explored in other trials of zoledronic acid and reanalyses of data from other bisphosphonate trials.
In conclusion, a once-yearly infusion of zoledronic acid during a 3-year period was associated with a significant and sustained decrease in the risk of vertebral, hip, and other fractures. In addition, the treatment had a favorable safety profile and was generally well tolerated. Given the relatively poor adherence to oral bisphosphonate therapy in clinical practice, annual infusion of zoledronic acid may provide a promising approach to reducing fracture risk.
Supported by Novartis Pharma.
Methods
Study Design
The Health Outcomes and Reduced Incidence with Zoledronic Acid Once Yearly (HORIZON) Pivotal Fracture Trial was an international, multicenter, randomized, double-blind, placebo-controlled trial involving postmenopausal women with osteoporosis. Patients were randomly assigned to receive either a 15-minute intravenous administration of zoledronic acid (5 mg) or placebo at baseline (day 0), at 12 months, and at 24 months. In addition, all patients received oral daily calcium (1000 to 1500 mg) and vitamin D (400 to 1200 IU). Patients were monitored for 3 years with quarterly telephone interviews and clinic visits at months 6, 12, 24, and 36.
The study was jointly designed by members of the steering committee and the sponsor. The sponsor had responsibility for data collection and quality control. An independent data and safety monitoring board met semiannually to oversee the conduct of the study and monitor the safety of patients. A copy of the study database was periodically transferred to the University of California, San Francisco (UCSF), for reports to the data and safety monitoring board. Analyses for publication were the joint responsibilities of representatives of the sponsor and investigators at UCSF. The original analyses were performed by the sponsor but were independently confirmed by investigators at UCSF. All the authors contributed to the writing of the article, and approval from a majority of the 13-member steering committee, which included 2 representatives of the sponsor, was required.
Patients
Postmenopausal women between the ages of 65 and 89 years were eligible for inclusion if they had a bone mineral density T score of -2.5 or less at the femoral neck, with or without evidence of existing vertebral fracture, or a T score of -1.5 or less, with radiologic evidence of at least two mild vertebral fractures or one moderate vertebral fracture. Previous use of oral bisphosphonates was allowed, with the duration of the washout period dependent on previous use (e.g., previous use of 48 weeks required 2 years of washout). Concomitant use of the following osteoporosis medications was allowed at baseline and during follow-up: hormone therapy, raloxifene, calcitonin, tibolone, tamoxifen, dehydroepiandrosterone, ipriflavone, and medroxyprogesterone. Patients were placed into one of two strata on the basis of whether they were taking osteoporosis medications at baseline. Patients in stratum 1 were not taking any osteoporosis medications at the time of randomization, whereas patients in stratum 2 were all taking an allowed medication. Patients who had previously taken bisphosphonates and met washout criteria could be randomly assigned to either stratum. Ineligibility criteria included any previous use of parathyroid hormone or sodium fluoride, use of anabolic steroids or growth hormone within 6 months before trial entry or oral or intravenous systemic corticosteroids within 12 months, and any previous use of strontium. Patients with a serum calcium level of more than 2.75 mmol per liter or less than 2.00 mmol per liter were ineligible, as were patients with a calculated creatinine clearance of less than 30.0 ml per minute at either of two baseline visits or urine dipstick results of more than 2+ for protein, without evidence of contamination or bacteriuria.
From February 2002 to June 2003, patients underwent randomization with the use of random permuted blocks within strata; the last closeout visit occurred on June 15, 2006. All patients provided written informed consent, and the local institutional review board at each center approved the protocol.
End Points
The primary end points were new vertebral fractures (in stratum 1) and hip fracture (in both strata). Secondary efficacy end points included any nonvertebral fracture, any clinical fracture, and clinical vertebral fracture. Other secondary end points were changes in bone mineral density at the total hip, femoral neck, and lumbar spine and changes in markers of bone resorption (serum C-telopeptide of type I collagen) and formation (bone-specific alkaline phosphatase and N-terminal propeptide of type I collagen). Data for premenopausal normative markers were based on 2.5th and 97.5th percentiles derived from the Os des Femmes de Lyon (OFELY) study12,13 (Delmas PD and Garnero P: personal communication). Height was measured with the use of a stadiometer, where available, at baseline and at months 12, 24, and 36.14 All investigators who performed end-point evaluations were unaware of patients' study-group assignments.
Efficacy Measurements
Spinal lateral radiographs were obtained at baseline and at 12, 24, and 36 months or early termination for patients in stratum 1 and at baseline and at 36 months or early termination for patients in stratum 2. Vertebrae from T4 to L4 were evaluated by an expert reader at a central imaging laboratory (Synarc) with the use of quantitative morphometry and standard methods.14 Incident morphometric vertebral fractures were defined as a reduction in vertebral height of at least 20% and 4 mm by quantitative morphometry, confirmed by an increase of one severity grade or more on semiquantitative analysis.14 Prevalent fracture at baseline was defined by a height ratio of at least 3 SD below the vertebra-specific mean height ratio on quantitative reading with semiquantitative confirmation.15,16
Clinical fracture reports were obtained from patients at each contact. Nonvertebral fracture reports required central confirmation, which was performed at the UCSF Coordinating Center. Evidence included either a radiologic or surgical procedure report or a copy of the radiograph. Excluded were fractures of the toe, facial bone, and finger and those caused by excessive trauma (assessed centrally as sufficient to cause fracture in a person without osteoporosis2). For clinical vertebral fractures, the community-obtained radiograph was compared with the baseline study radiograph by a central reader at Synarc, and semiquantitative confirmation was required.
Dual-energy x-ray absorptiometry of the hip was performed at baseline and at months 6, 12, 24, and 36. Investigators and coordinators were unaware of study-group assignments of patients regarding the results of follow-up scans. Bone loss was monitored centrally and if a patient's loss of bone mineral density at the total hip exceeded 8% at 1 year or 10% at 2 years, the site was notified, and the patient counseled regarding alternative treatment options. Measurements of bone mineral density at the lumbar spine were obtained for a subgroup of patients. All measurements of bone mineral density were corrected for site variations.
Levels of serum C-telopeptide of type I collagen and bone-specific alkaline phosphatase were measured in serum samples obtained after an overnight fast in a subgroup of about 600 patients from selected clinical sites. An additional cohort underwent measurement of levels of N-terminal propeptide of type I collagen from samples shipped to the central laboratory at ambient temperature. Samples obtained at all time points for each patient were analyzed in a single batch, whenever possible, at Synarc.
Adverse Events
Safety was assessed by the recording of all adverse events and serious adverse events and by physical examination, regular measurement of vital signs, and regular monitoring of hematologic, blood-chemical, and urinary values. Adverse events were categorized according to codes used in the Medical Dictionary for Regulatory Activities (MedDRA).17 The five most commonly reported symptoms that occurred within 3 days after an infusion of a study drug (post-dose symptoms) �\ pyrexia, influenza-like symptoms, myalgia, headache, and arthralgia �\ were analyzed individually and grouped.
To assess renal safety, serum creatinine was measured in a subgroup of 5035 patients 9 to 11 days after each infusion. A significant increase was defined as a rise of more than 0.5 mg per deciliter (44 ��mol per liter) in the serum creatinine level, as compared with the baseline level before the first infusion. A total of 559 patients underwent 12-lead electrocardiography before and 9 to 11 days after the third infusion. Patients who were taking medications associated with QT-interval prolongation were excluded from this substudy. Adjudication or expert review was performed for several categories of adverse events, including ocular events, osteonecrosis of the jaw, hypocalcemia, renal events, incomplete fracture healing, cardiovascular events, hip and knee osteonecrosis, and death. Each adjudication committee created a set of predefined search terms on the basis of codes from MedDRA and the World Health Organization Drug Reference List. The adverse-event database was then searched for these terms. Investigators at each clinical center collected medical documentation for the cases. This documentation was forwarded to the expert panels, which performed event adjudication while members were unaware of study-group assignments. Maxillofacial events that were possibly associated with a diagnosis of osteonecrosis of the jaw were adjudicated on the basis of a definition of the disorder as the presence of exposed bone for more than 6 weeks.
Statistical Analysis
The plan for the analysis of data, which was developed before unblinding, prespecified all statistical analyses. Efficacy analyses included all patients who had undergone randomization, except for 29 patients from a site whose participation had been terminated during the study. Patients who had received at least one infusion were included in safety analyses (Figure 1).
Completion of follow-up was defined as receipt of a baseline infusion at the time of randomization and attendance at a study closeout visit. The primary reasons that patients in both study groups did not complete follow-up were adverse events, withdrawal of consent, loss to follow-up, and death. A total of 29 patients (14 in the zoledronic-acid group and 15 in the placebo group) were excluded from all analyses because the participation of their clinical center was terminated, owing to issues associated with data reliability. In addition, 22 patients in both groups who did not receive a study drug at baseline were excluded from the safety analysis but were included in the efficacy analysis.
The incidence of vertebral fracture (stratum 1) included patients who had undergone radiography at baseline and at least once during follow-up. An incident fracture was identified if at least one follow-up radiograph met the criteria for incident fracture. Results are presented as the relative risk and 95% confidence interval (CI).
Clinical fractures (including hip fracture) were analyzed with the use of a proportional-hazards model stratified for the study stratum, with the relative hazard reported and the cumulative proportion of patients with fractures estimated by means of Kaplan-Meier analysis. Follow-up time was defined as the time from randomization to the first relevant fracture, the last study visit, or the time of death, whichever occurred first. Changes in bone mineral density were compared with the use of analysis of variance adjusted for stratum and region. Changes in biochemical markers were compared by means of analysis of covariance (ANCOVA) (loge ratio of the post-baseline value to the baseline value) adjusted for stratum, center, and baseline value. The incidence of safety events was compared with the use of Fisher's exact test.
One interim analysis of the two primary end points was conducted for the data and safety monitoring board and the final significance levels were adjusted accordingly to P=0.0496 for vertebral fracture and P=0.0406 for hip fracture. For all other tests, a P value of 0.05 or less was considered to indicate statistical significance. All reported P values are two-sided. No adjustments were made for multiple comparisons of the safety end points.
The study had a power of 90% (with a two-sided alpha of 0.05) to detect a 50% reduction in morphometric vertebral fractures in the zoledronic-acid group, assuming an annual incidence of 1.9% in the placebo group, with 2252 patients in stratum 1 (the number that was originally projected). With 7400 patients, the log-rank test had a power of 90% to detect a 50% reduction in hip fractures, assuming a 3-year fracture rate of 1.8% in the placebo group.
|
|
| |
| |
|
|
|