| |
Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-1: 24-week results from a randomised, double-blind, placebo-controlled trial
|
| |
| |
The Lancet July 7, 2007
Dr Jose Valdez Madruga MD a , Pedro Cahn MD b, Beatriz Grinsztejn MD c, Prof Richard Haubrich MD d, Jacob Lalezari MD e, Anthony Mills MD f, Prof Gilles Pialoux MD g, Timothy Wilkin MD h, Monika Peeters MSc i, Johan Vingerhoets PhD i, Goedele de Smedt MD i, Lorant Leopold MD j, Roberta Trefiglio PharmD j and Brian Woodfall MD i, on behalf of the DUET-1 study group
Summary
Background
Antiretroviral agents active against drug-resistant HIV-1 are needed for treatment-experienced patients. The aim of this trial was to assess the efficacy, safety, and tolerability of TMC125 (etravirine), a non-nucleoside reverse transcriptase inhibitor (NNRTI).
Methods
DUET-1 is a continuing, multinational randomised, double-blind, placebo-controlled, phase III trial. Treatment-experienced adult patients with virological failure on stable antiretroviral therapy, documented genotypic evidence of NNRTI resistance, viral load over 5000 copies per mL, and three or more primary protease inhibitor mutations were randomly assigned to receive 200 mg TMC125 or placebo twice daily. All patients also received darunavir with low-dose ritonavir and investigator-selected nucleoside reverse transcriptase inhibitors. Enfuvirtide use was optional. The primary endpoint was a confirmed viral load below 50 copies per mL at week 24 (FDA time-to-loss of virological response algorithm). Analyses were done by intention to treat. This trial is registered with ClinicalTrials.gov, with the number NCT00254046.
Findings
612 patients were randomised and treated (304 in the TMC125 group, 308 in the placebo group). By week 24, 42 (14%) patients in the TMC125 group and 56 (18%) in the placebo group had discontinued, mainly due to virological failure. At week 24, 170 (56%) patients in the TMC125 group and 119 (39%) patients in the placebo group achieved a confirmed viral load of less than 50 copies per mL (difference in response rates 17%; 95% CI 9-25; p=0·005). Most adverse events were mild or moderate in severity. The type and incidence of adverse events, including neuropsychiatric events, seen with TMC125 were generally comparable with placebo, with the exception of rash (61 [20%] patients on TMC125 vs 30 [10%] on placebo) and diarrhoea (36 [12%] patients on TMC125 vs 63 [20%] on placebo).
Interpretation
In treatment-experienced patients with NNRTI resistance, treatment with TMC125 achieved better virological suppression at week 24 than did placebo. The safety and tolerability profile of TMC125 was generally comparable with placebo.
Introduction
The current therapeutic goal for treatment-experienced HIV-1-infected patients is maximum virological suppression (viral load <50 copies per mL),1 a target previously reserved for patients with little or no history of antiretroviral therapy. However, therapeutic options for treatment-experienced patients are still limited by toxicity and the development of antiretroviral drug resistance. Single aminoacid substitutions in HIV-1 reverse transcriptase can lead to broad non-nucleoside reverse transcriptase inhibitor (NNRTI) class cross-resistance2,3 and limit the use of NNRTIs in this patient population. In vitro, TMC125 (etravirine), a NNRTI, is highly active against both wild-type and NNRTI-resistant HIV strains and has a higher genetic barrier to the development of resistance than currently available NNRTIs.4,5 In phase IIb trials in treatment-experienced patients, including those infected with virus resistant to NNRTIs and protease inhibitors (PIs), TMC125 was active against HIV resistant to currently available NNRTIs, with a similar tolerability profile to that of the control group.6-8
DUET-1 is a phase III trial which, together with the DUET-2 trial,9 is designed to assess the long-term efficacy, tolerability, and safety of TMC125 in treatment-experienced HIV-1-infected patients. This paper reports the protocol-specified week 24 primary analysis.
Methods
Patients
This continuing phase III, randomised, double-blind, placebo-controlled trial was done in Argentina, Brazil, Chile, France, Mexico, Panama, Puerto Rico, Thailand, and the USA. Patients were recruited between Nov 10, 2005, and July 18, 2006. Patients will have the option to extend the initial 48-week treatment period by a further 48 weeks.
To be considered for inclusion, patients were aged 18 years or over with documented HIV-1 infection and receiving a stable antiretroviral regimen initiated at least 8 weeks before screening. Eligible patients had plasma viral load (HIV-1 RNA) over 5000 copies per mL, three or more primary PI mutations10 at screening, and at least one NNRTI resistance-associated mutation at screening or from a historical genotype. NNRTI resistance-associated mutations were defined as Ala98Gly, Leu100Ile, Lys101Glu/Pro/Gln, Lys103His/Asn/Ser/Thr, Val106Ala/Met, Val108Ile, Glu138Gly/Lys/Gln, Val179Ile/Phe/Gly, Tyr181Cys/Ile/Val, Tyr188Cys/His/Leu, Gly190Ala/Glu/Ser, Pro225His, Phe227Cys, Met230Ile/Leu, Pro236Leu, Lys238Asn/Thr, and Tyr318Phe.10,11 Patients were excluded if they had a life expectancy of less than 6 months, any currently active AIDS-defining illness, acute viral hepatitis, or, if female, were pregnant, breastfeeding, or of child-bearing potential and not using adequate contraception. Individuals with chronic hepatitis B or C infection were not excluded provided that concentrations of aspartate aminotransferase and alanine aminotransferase were less than five times the upper limit of normal and treatment was not deemed to be necessary by the investigator. Investigational drugs (other than darunavir) and agents with potential drug interactions were prohibited during the trial.
Written informed consent was obtained from all patients. The protocol was reviewed and approved by the appropriate institutional review boards or ethics committees and health authorities and the trial was done in accordance with the Declaration of Helsinki.
Procedures
Patients were randomised in a 1:1 fashion to receive either 200 mg TMC125 or placebo (twice daily after a meal) in a double-blinded fashion, using interactive voice or web-based response systems based on a validated adaptive minimisation technique12 with biased coin assignment. The TMC125 200 mg twice daily dose regimen, using two 100 mg tablets twice daily of the new formulation, results in exposure comparable with 800 mg twice daily (the dose selected after phase IIb trials) of the earlier phase II formulation (unpublished data).13 Randomisation was stratified for enfuvirtide use in the background regimen (no use, re-use, or de-novo use), previous darunavir use (used or not), and screening plasma viral load (<30 000 or ≥30 000 copies per mL). Before randomisation, investigators selected a background regimen to be received by each patient during the treatment period, including nucleoside reverse transcriptase inhibitors (NRTIs; selected based on screening genotypic resistance [vircoTYPE HIV-1; Virco BVBA, Belgium] and treatment history) and optional enfuvirtide. All patients also received the PI darunavir (TMC114; Tibotec, Belgium), with low-dose ritonavir as part of the background antiretroviral regimen (600 mg darunavir plus 100 mg ritonavir twice daily). Since darunavir-ritonavir has shown antiviral efficacy in treatment-experienced HIV-1-infected patients,14 the use of darunavir-ritonavir by all patients ensured that the trial population received at least one potentially active agent (other than TMC125) with efficacy in this population. Additionally, administration of these two investigational agents was deemed to be appropriate because the use of at least two new antiretrovirals in a regimen has the potential to improve results in treatment-experienced patients.15 Regimens could not be changed except for tolerability or toxicity reasons.
All endpoints were predefined in the protocol. The primary endpoint was the proportion of patients achieving a confirmed viral load less than 50 copies per mL at week 24 (intention-to-treat [ITT] analysis, time-to-loss of virological response [TLOVR] imputation algorithm16). Secondary endpoints were the proportion of patients achieving a viral load less than 400 copies per mL, change in viral load from baseline, change in CD4 cell count from baseline, and safety and tolerability. The primary efficacy endpoint was also analysed according to baseline viral load of under 100 000 copies per mL and 100 000 copies per mL or more, considered to be clinically relevant in this treatment-experienced population. Results from the analyses of additional secondary endpoints (HIV-1 genotype and drug susceptibility changes, population pharmacokinetics, assessment of the pharmacokinetic-pharmacodynamic relation between TMC125 and darunavir, and assessment of global health status based on the Functional Assessment of HIV Infection questionnaire) will be reported elsewhere.
Viral load was measured with the Amplicor HIV-1 monitor ultrasensitive assay (version 1.5; Roche Diagnostics, Basel, Switzerland); immunological change was assessed by CD4 counts (absolute and percentage). Phenotype was determined with Antivirogram and genotype with vircoTYPE HIV-1 (both Virco BVBA, Belgium).
An independent data and safety monitoring board monitored and assessed adverse events and laboratory abnormalities on a scheduled basis, and regularly reviewed available safety and efficacy data. Safety assessments and clinical laboratory tests included the reporting of adverse events and HIV-related events, physical examinations, electrocardiographs, vital signs, urinalysis, haematology, and biochemistry (patients fasted for at least 10 h before blood sampling). Patients were withdrawn for pregnancy, grade 3 or 4 allergic events or skin reactions, grade 4 adverse events or confirmed laboratory abnormalities (excluding adverse events deemed by the investigator to be doubtfully related or unrelated to trial medication and protocol-defined grade 4 increases in glucose and triglyceride concentrations), clinical hepatitis, and disallowed alterations in medication.
Virological failure leading to treatment discontinuation was defined as viral load reduction of less than 0·5 log10 copies per mL from baseline at week 8 or less than 1·0 log10 copies per mL at week 12 or beyond (one subsequent confirmatory result was required in both cases). Loss of virological response was defined as plasma viral load of more than 0·5 log10 copies per mL above the nadir in two consecutive measurements after a minimum of 12 weeks. Patients experiencing a viral rebound at or beyond week 24, or failing to achieve a reduction in viral load of more than 1·0 log10 copies per mL from baseline at week 24, were given the opportunity to receive TMC125 plus darunavir-ritonavir in an open-label rollover trial (TMC125-C217; ClinicalTrials.gov reference NCT00359021). Investigators will continue to provide survival data for all withdrawn patients every 6 months until the end of the trial.
Statistical analysis
All analyses were done on the ITT population (ie, all randomised patients who had received at least one dose of trial medication, irrespective of protocol compliance or ineligibility). The primary efficacy analysis was done when all patients had reached week 24 or discontinued earlier (database cutoff date Feb 9, 2007).
The sample size was based on the 24-week interim results of darunavir-ritonavir in the phase IIb POWER trials.17 Response in the placebo group was estimated at 35% (when enfuvirtide was re-used or not used) and 60% (when enfuvirtide was used de novo), and response in the TMC125 group was estimated at 55% (when enfuvirtide was re-used or not used) and 60% (when enfuvirtide was used de novo). To show that TMC125 was better than placebo with at least 95% power, the population using enfuvirtide de novo was limited to 40% and 300 patients were required for each treatment group. All statistical tests were interpreted at the two-sided 5% significance level.
For the primary efficacy endpoint, the TLOVR imputation algorithm was applied,16 incorporating non-completer=failure (NC=F) imputation in the case of patient discontinuation and last observation carried forward for missing data in other cases. Two consecutive viral load values were needed for confirmation of response or loss of response. The difference in virological response rates between the treatment groups at week 24 (controlling for the stratification factors) was assessed with the Cochran-Mantel-Haenszel test. The effect of adding TMC125 to antiretroviral therapy was expected to be different between patients who re-used or did not use enfuvirtide and patients who used the drug de novo. Based on guidance from the US Food and Drug Administration at the protocol development stage, a test for a significant statistical interaction effect between TMC125 and enfuvirtide use was done with the Breslow-Day test for the homogeneity of odds ratios before the main statistical analysis. The presence of a significant statistical interaction between TMC125 and enfuvirtide use (p<0·20) required separate Cochran-Mantel-Haenszel tests to be done by enfuvirtide strata, with a Hochberg multiplicity correction applied. The overall effect of TMC125 versus placebo was also estimated across both enfuvirtide strata (ie, enfuvirtide re-used or not used; enfuvirtide used de novo) with a logistic regression model. To assess the effect of baseline characteristics and resistance parameters on virological response, logistic regression models were applied. Enfuvirtide was considered to be active if used de novo; darunavir was considered to be active if darunavir fold change in 50% effective concentration (FC) was less than 10;18 and NRTIs were defined as active according to baseline phenotypic analysis if FC was below the cutoffs used in the Antivirogram assay.
For the changes in plasma viral load and CD4 count versus baseline at all time points, the least square mean of the difference between the TMC125 and placebo groups and the two-sided 95% CI were estimated with an analysis of covariance model. A generalised linear mixed effects model was used to describe the plasma viral load changes over time. The TLOVR algorithm was applied to the analysis of the secondary virological response parameters. A predefined comparative analysis of the incidence of rash and neuropsychiatric adverse events between the TMC125 and placebo groups was done with the Fisher's exact test.
This trial is registered with ClinicalTrials.gov, with the number NCT00254046.
Role of the funding source
The study sponsor was involved in the trial design and conduct. Data were collected and analysed by the sponsor. All authors had full access to the 24-week trial data and the corresponding author had final responsibility to submit the manuscript for publication.
Results
1220 patients were screened; of these, 612 were randomised and treated (304 in the TMC125 group, 308 in the placebo group). All had reached week 24 or discontinued earlier at the time of analysis (figure 1). 42 (14%) patients in the TMC125 group and 56 (18%) in the placebo group discontinued the trial, mainly because of virological failure. At the time of analysis, median treatment duration was 26·6 (range 0·9-60·3) weeks in the TMC125 group and 26·6 (range 2·7-55·3) weeks in the placebo group.
Demographics and baseline disease characteristics were similar between the TMC125 and placebo treatment groups (table 1). Overall, 84 (14%) patients were receiving NNRTIs during the screening period. Of the 601 patients with available baseline phenotypic sensitivity data (299 in the TMC125 group, 302 in the placebo group), 323 (54%) were resistant to all currently available NRTIs (161 in the TMC125 group, 162 in the placebo group), 178 (30%) were sensitive to one (82 in the TMC125 group, 96 in the placebo group), and 100 (17%) were sensitive to two or more (56 in the TMC125 group, 44 in the placebo group). The two treatment groups were balanced in terms of both the number of NRTIs and individual NRTIs patients used in their background regimen. Enfuvirtide was used during the treatment period by 121 (40%) patients in the TMC125 group and 127 (41%) in the placebo group. Most patients (388 [65%]) were sensitive to darunavir (FC<10), which was the most frequently fully active PI of the currently available PIs. The three most prevalent NNRTI resistance-associated mutations at baseline were Lys103Asn (198 [32%] patients), Tyr181Cys (180 [29%] patients), and Gly190Ala (163 [27%] patients). The occurrence of these mutations was balanced between the two groups.
A confirmed viral load of less than 400 copies per mL was achieved by 224 (74%) patients in the TMC125 group and 158 (51%) patients in the placebo group at week 24 (difference in response rates 22%, 95% CI 15-30; p=0·0001; figure 2). Analysis of response by the number of active agents in the background regimen showed that greater proportions of patients in the TMC125 group achieved viral loads below 50 copies per mL and less than 400 copies per mL than in the placebo group, irrespective of the number of active agents (figure 2). A viral load below 50 copies per mL was achieved by 21 (47%) of the 45 patients in the TMC125 group with no active agents in their background regimen, compared with four (9%) of 46 patients in the placebo group.
The mean change in viral load from baseline in the TMC125 group was -2·41 (SD 1·28) log10 copies per mL, compared with -1·70 (1·49) log10 copies per mL in the placebo group (p<0·0001). CD4 cell count increased by a mean of 89 (93·65) cells per μL in the TMC125 group and 64 (91·41) cells per μL in the placebo group (p=0·0002; figure 2).
CDC category C AIDS-defining illnesses or death were reported in eight (3%) patients in the TMC125 group and 21 (7%) patients in the placebo group (p=0·1633 in logistic regression analysis). CDC category C AIDS-defining illnesses were reported in five (2%) patients in the TMC125 group and 20 (7%) patients in the placebo group (p=0·0384 in logistic regression analysis). There were four (1%) deaths in the TMC125 group and eight (3%) in the placebo group (p=0·7999).
A significant statistical interaction effect was identified between TMC125 and enfuvirtide use (p=0·0460). Further statistical analyses comparing efficacy between the TMC125 and placebo groups were therefore based on two enfuvirtide subgroups: patients who re-used or did not use enfuvirtide, and patients who used enfuvirtide de novo.
Of the patients randomised to the TMC125 group, 74 (24%) patients used enfuvirtide de novo, 47 (16%) re-used enfuvirtide, and 183 (60%) did not use the drug. In the placebo group, 79 (26%) used enfuvirtide de novo, 48 (16%) patients re-used enfuvirtide, and 181 (59%) did not use the drug. Of the patients re-using or not using enfuvirtide, more patients in the TMC125 group (126 patients [55%]) reached the primary endpoint than did those in the placebo group (75 patients [33%]; difference in response rate 22%, 95% CI 13-31; p<0·0001; figure 3). Similarly, there were more patients in the TMC125 group achieving a viral load below 400 copies per mL (162 [70%] patients) than in the placebo group (100 [44%] patients; difference in response rate 27%, 18-36; p<0·0001; figure 3). The proportion of patients achieving a viral load below 50 copies per mL was generally greater in the TMC125 group than in the placebo group irrespective of the baseline number of NNRTI resistance-associated mutations (figure 3). More patients re-using or not using enfuvirtide in the TMC125 group achieved a viral load below 50 copies per mL, irrespective of baseline darunavir FC, than did those in the placebo group (figure 3).
In the subgroup of patients that re-used or did not use enfuvirtide, AIDS-defining illnesses or deaths were recorded in five (2%) patients in the TMC125 group and 18 (8%) patients in the placebo group (p=0·0081 in logistic regression analysis). AIDS-defining illnesses were reported in three (1%) patients in the TMC125 group and 17 (7%) patients in the placebo group (p=0·0026 in logistic regression analysis). There were two (1%) deaths in the TMC125 group and seven (3%) in the placebo group (p=0·1582).
Of the patients who used enfuvirtide de novo, the primary endpoint was reached by 44 (59%) patients in the TMC125 group and 44 (56%) patients in the placebo group (difference in response rate 4%, 95% CI -12 to 20; p=0·7935; figure 3). 62 (84%) patients in the TMC125 group and 58 (73%) in the placebo group achieved a viral load below 400 copies per mL (difference in response rate 10%, -3 to 23; p=0·1218; figure 3). Analysis by number of baseline NNRTI resistance-associated mutations showed that, in general, more patients reached the primary endpoint in the TMC125 group than did those in the placebo group (figure 3). Response rates were much the same between the TMC125 and placebo groups for patients receiving enfuvirtide de novo with darunavir FC below 10, and were numerically higher in the TMC125 group than in the placebo group for those with FC values of 10-40 and over 40 (figure 3).
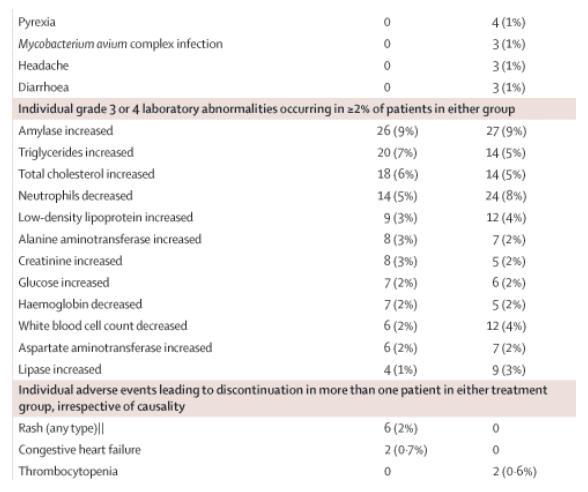
The safety analysis included all available data from all patients up to the database cutoff date. Overall, 282 (93%) patients in the TMC125 group and 287 (93%) patients in the placebo group reported at least one adverse event during the treatment period. The frequencies of adverse events were comparable between the TMC125 and placebo groups and were mostly grade 1 or 2 in severity (table 2). The frequencies of individual grade 3 or 4 adverse events, irrespective of causality, in the TMC125 group were comparable with the placebo group; no consistent pattern of individual grade 3 or 4 adverse events was seen. Overall, 35 (12%) patients in the TMC125 group and 62 (20%) in the placebo group reported at least one serious adverse event (p=0·0039 for TMC125 vs placebo), most of which were typical of conditions that occur commonly in HIV-1-infected patients. No serious adverse events were recorded in more than 1% of patients in the TMC125 group; no consistent pattern of type, frequency, or relation to TMC125 was seen. Adverse events leading to permanent treatment discontinuation were seen in 16 (5%) patients in both the TMC125 and the placebo group. Rash (any type) was seen in 61 (20%) patients in the TMC125 group and 30 (10%) patients in the placebo group (p<0·0001 for TMC125 vs placebo). In the TMC125 group, most cases of rash were grade 1 or 2 (mild or moderate) in severity. Grade 3 rash was recorded in four (1%) patients; no grade 4 rashes were reported. The grade 1 or 2 rashes were maculopapular in nature, and tended to occur within the first few weeks of treatment (median onset 11 days) and resolved with continued treatment (median duration 12 days). Rash led to trial discontinuation in six (2%) patients in the TMC125 group (four during the treatment period and two during follow-up) and none in the placebo group (table 2). In the TMC125 group, the proportion of patients with rash was higher in female (14 [34%] of 41 women) than in male participants (47 [18%] of 263 men; p=0·0192), although there was no sex difference in severity or frequency of treatment discontinuation due to rash.
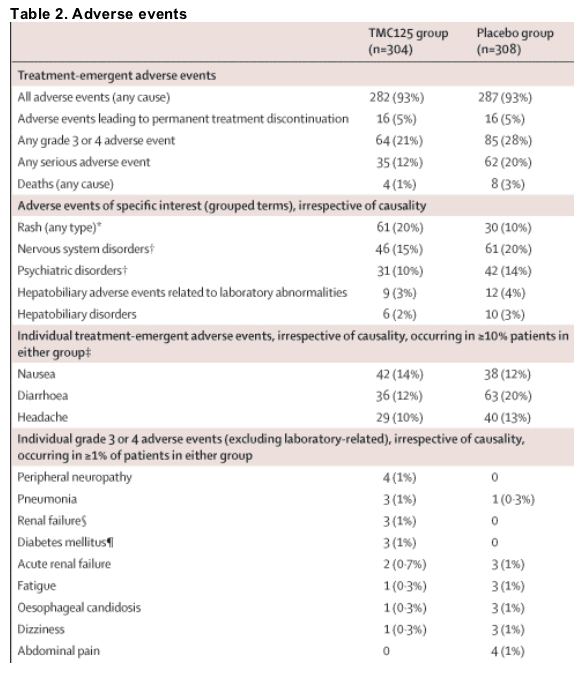
Neuropsychiatric adverse events (including nervous system, psychiatric, and certain eye disorders) were mostly grade 1 or 2 in severity and were noted in 69 (23%) patients in the TMC125 group and 94 (31%) patients in the placebo group (p=0·249 for TMC125 vs placebo). One (0·3%) patient in the TMC125 group and nine (3%) patients in the placebo group experienced a grade 3 neuropsychiatric adverse event; no grade 4 cases were recorded. Individual nervous system adverse events and psychiatric adverse events in the TMC125 group were mostly mild or moderate in severity and were no different from placebo in frequency.
No clinically relevant changes in laboratory parameters were noted over the treatment period, with no differences between the TMC125 and placebo groups. In the overall population, TMC125 was not found to have any clinically relevant effect on lipid concentrations, including triglycerides, compared with placebo. The incidence of grade 3 and 4 laboratory abnormalities (including lipid, hepatic, and pancreatic) was comparable between the treatment groups.
Four (1%) patients in the TMC125 group and eight (3%) patients in the placebo group died due to adverse events that began during the treatment period. The causes of death in the four patients of the TMC125 group were: congestive cardiac failure, central line infection and septic shock; cardiogenic shock in the setting of pneumonia and congestive cardiac failure; renal impairment (deemed to be tenofovir-related), respiratory tract infection and respiratory failure; and sudden death due to artherosclerotic and hypertensive cardiovascular disease. None of these deaths was deemed to be related to TMC125.
Discussion
At week 24, the primary efficacy endpoint of confirmed viral load below 50 copies per mL was achieved by a significantly greater proportion of treatment-experienced patients receiving TMC125 than those receiving placebo. Additional benefits of TMC125 over placebo were seen for secondary endpoints including viral load below 400 copies per mL, change in viral load from baseline, and change in CD4 cell count from baseline.
The occurrence of AIDS-defining illnesses and deaths at week 24 was not significantly different between the TMC125 and placebo groups in the overall population but did reach significance in the subgroup of patients that re-used or did not use enfuvirtide. Most treatment-emergent adverse events were mild or moderate in severity, and the occurrence of serious or grade 3 or 4 events, and of events leading to discontinuation, was similar in both groups. Although rash (any type) occurred more frequently in the TMC125 group than in the placebo group, cases were generally mild to moderate in severity, infrequently led to discontinuation, and generally resolved with continued treatment. These observations are similar to those of a recent phase IIb trial.7 Additionally, the nature, frequency, and severity of hepatic, neuropsychiatric, and lipid-related adverse events did not differ between the TMC125 and placebo groups in the present trial.
The results of this trial show that, at 24 weeks, TMC125 has antiviral activity in patients with NNRTI resistance. The findings are consistent with the in-vitro activity of TMC125 against NNRTI-resistant HIV strains.4,5 The virological responses noted in the TMC125 group were better than those of the placebo group, despite all patients having at least one documented NNRTI resistance-associated mutation and an extensive treatment history. Other currently available NNRTIs would not generally be expected to provide a virological response in such patients, since the presence of a single NNRTI resistance-associated mutation has been shown to cause cross-resistance.3 TMC125 therefore offers a new treatment option within the NNRTI class, with the results of the trial also indicating that sequential use of TMC125-ie, use of TMC125 after another NNRTI-is probably effective even in the presence of NNRTI resistance. The results of this trial are also lent support by the findings of the DUET-2 trial.9
The magnitudes of the efficacy responses seen in the placebo group reflect the use by all patients of darunavir, to which most patients were sensitive compared with other currently available PIs. These responses are consistent with pooled 24-week results of darunavir-ritonavir in the POWER 1 and 2 trials in treatment-experienced patients.14 Current recommendations state that at least two active agents (based on resistance testing and treatment history)15,20,21 should be used to give optimum virological suppression in treatment-experienced patients. As data from clinical trials of new antiretroviral agents become available,22-25 combining active agents in a regimen will help to provide virological suppression comparable with that seen in treatment-naive patients. Irrespective of the number of active agents in the background regimen, more patients in the TMC125 group than in the placebo group achieved a viral load below 400 or below 50 copies per mL, although, as would be expected, the magnitude of difference in response between the two treatment groups decreased as the number of active agents in the background regimen increased. For patients with no active agents in their background regimen, responses in both treatment groups were probably influenced by darunavir-ritonavir (since darunavir activity might have been retained in some patients with darunavir FC values between 10 and 40); however, the magnitude of virological response in the TMC125 group indicates a considerable added benefit of TMC125 in this patient subgroup, which is encouraging in a population where many patients have few remaining active treatment options.
The addition of TMC125 to de-novo enfuvirtide seemed to increase virological response in patients with some degree of resistance to darunavir (FC>10), underscoring the need for at least two active agents to be included in a regimen. Further studies are needed to identify which subgroups of patients require three or more active drugs in a regimen and whether this would reduce the risk of viral rebound.
One of the limitations of the trial is that the PI component of the background regimen was fixed with darunavir-ritonavir. Since TMC125 cannot be coadministered with the PI tipranavir,26 darunavir was considered the best PI option for this population with extensive PI resistance. This decision is validated by the responses seen in the placebo group. Although the trial does not provide information on TMC125 combined with other PIs, one should be able to extrapolate the data to other boosted PIs. The data are supported by a phase II study of TMC125 combined with lopinavir-ritonavir.27 Another limitation is that the protocol allowed historical evidence of NNRTI resistance-associated mutations, and 10% of all patients had no detectable NNRTI resistance-associated mutations at baseline. The trial therefore consisted of patients with detectable baseline NNRTI resistance as well as those with undetectable baseline-but archived-NNRTI resistance. However, it is not expected that currently available NNRTIs would provide virological suppression in either of these patient subgroups, while the added benefit of TMC125 was apparent irrespective of subgroup.
Preliminary analyses of data from the DUET trials have shown that the concurrent presence of three or more of 13 specific NNRTI mutations (from Val90Ile, Ala98Gly, Leu100Ile, Lys101Glu, Lys101Pro, Val106Ile, Val179Asp, Val179Phe, Tyr181Cys, Tyr181Ile, Tyr181Val, Gly190Ala, and Gly190Ser) was associated with decreased virological response to TMC125.28 Longer-term results from the ongoing DUET studies will continue to establish the role and provide further guidance for the use of TMC125 in this patient population.
|
|
| |
| |
|
|
|