| |
Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV-infected patients in TITAN: a randomised controlled phase III trial
|
| |
| |
The Lancet July 7, 2007; 370:49-58
Pictures of tables are imbedded in this report.
Dr Jose Valdez Madruga MD a , Daniel Berger MD b, Marilyn McMurchie MD c, Fredy Suter MD d, Denes Banhegyi MD e, Kiat Ruxrungtham MD f, Dorece Norris MD g, Eric Lefebvre MD h, Marie-Pierre de Bethune PhD i, Frank Tomaka MD j, Martine De Pauw PharmD i, Tony Vangeneugden MSc i and Sabrina Spinosa-Guzman MD i, on behalf of the TITAN study group
Summary
Background
The protease inhibitor darunavir has been shown to be efficacious in highly treatment-experienced patients with HIV infection, but needs to be assessed in patients with a broader range of treatment experience. We did a randomised, controlled, phase III trial (TITAN) to compare 48-week efficacy and safety of darunavir-ritonavir with that of lopinavir-ritonavir in treatment-experienced, lopinavir-naive patients.
Methods
Patients received optimised background regimen plus non-blinded treatment with darunavir-ritonavir 600/100 mg twice daily or lopinavir-ritonavir 400/100 mg twice daily. The primary endpoint was non-inferiority (95% CI lower limit for the difference in treatment response -12% or greater) for HIV RNA of less than 400 copies per mL in plasma at week 48 (per-protocol analysis). TITAN (TMC114-C214) is registered with ClinicalTrials.gov, number NCT00110877.
Findings
Of 595 patients randomised and treated, 187 (31%) were protease inhibitor naive; 476 of 582 (82%) were susceptible to four or more protease inhibitors. At week 48, significantly more darunavir-ritonavir than lopinavir-ritonavir patients had HIV RNA of less than 400 copies per mL (77% [220 of 286] vs 68% [199 of 293]; estimated difference 9%, 95% CI 2-16). Fewer virological failures treated with darunavir-ritonavir than with lopinavir-ritonavir developed primary protease inhibitor mutations (21% [n=6] vs 36% [n=20]) and nucleoside analogue-associated mutations (14% [n=4] vs 27% [n=15]). Safety data were generally similar between the groups; grade 3 or 4 adverse events occurred in 80 (27%) darunavir-ritonavir and 89 (30%) lopinavir-ritonavir patients.
Interpretation
In lopinavir-naive, treatment-experienced patients, darunavir-ritonavir was non-inferior to lopinavir-ritonavir treatment in terms of our virological endpoint, and should therefore be considered as a treatment option for this population.
Results
Of 785 patients screened, 595 were randomised and treated (298 with darunavir-ritonavir and 297 with lopinavir-ritonavir) and were included in the ITT analysis. Data from 579 patients (286 in the darunavir-ritonavir and 293 in the lopinavir-ritonavir arm) were analysed in the per-protocol population to assess non-inferiority of virological response at week 48. All ITT patients were included in the safety analysis. At the time of this analysis, 53 patients (18%) in the lopinavir-ritonavir group had switched to the new tablet formulation and 38 patients (6%; 37 in the lopinavir-ritonavir group and one in the darunavir-ritonavir group) had entered the rollover period.
Overall, 25% (n=148) of randomised patients discontinued treatment (including 7% [n=41] due to an adverse event and 6% [n=38] due to virological failure). Figure 1 shows the overall trial profile. More patients withdrew from the lopinavir-ritonavir group than from the darunavir-ritonavir group, with 11% of patients in the lopinavir-ritonavir group discontinuing treatment due to virological failure compared with 1% in the darunavir-ritonavir group. The incidence of discontinuations due to an adverse event was low and was similar between treatment groups.
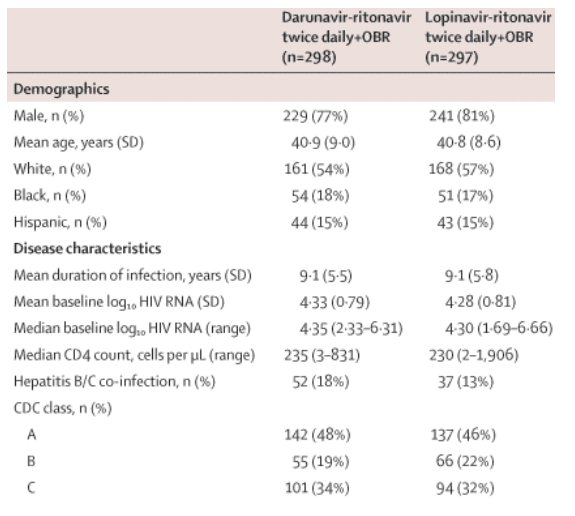
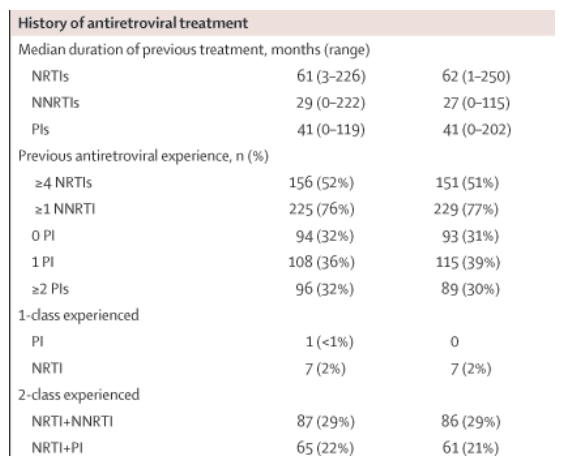
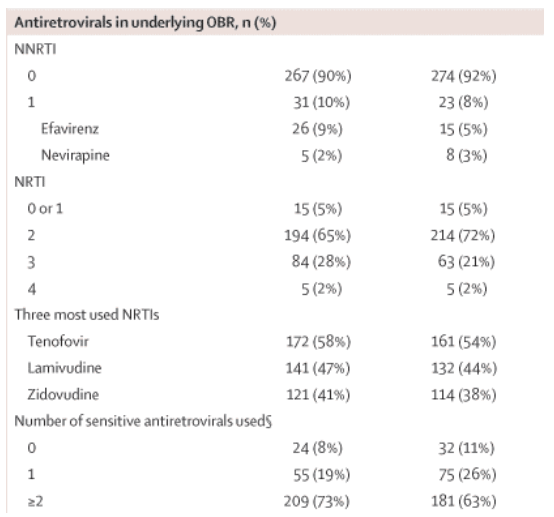
Baseline characteristics were well balanced between the two groups (table 1, see table below). A total of 109 patients (18%) had a baseline CD4 count of less than 100 cells per μL (55 [19%] darunavir-ritonavir, 54 [18%] lopinavir-ritonavir). The groups had similar numbers of patients on structured treatment interruption at screening, a reflection of clinical practice at the time of enrolment in the countries involved. At screening, the mean number of antiretroviral agents previously used by patients was 5·7 (mean numbers of protease inhibitors 1·2, NRTIs 3·6, and NNRTIs 0·9). The most frequently previously used antiretrovirals at screening were: protease inhibitors-indinavir (35%), nelfinavir (28%) and saquinavir (24%); NRTIs-lamivudine (93%), zidovudine (80%), and stavudine (60%); NNRTIs-efavirenz (50%) and nevirapine (42%).
The mean duration of study treatment was similar in both groups (53·5 weeks [SD 17·6] for darunavir-ritonavir and 51·5 weeks [17·4] for lopinavir-ritonavir). One patient in the lopinavir-ritonavir group and two in the darunavir-ritonavir group had been previously treated with lopinavir. All other patients had no previous exposure to either drug. The rates of previous use of other protease inhibitors were similar between the groups.
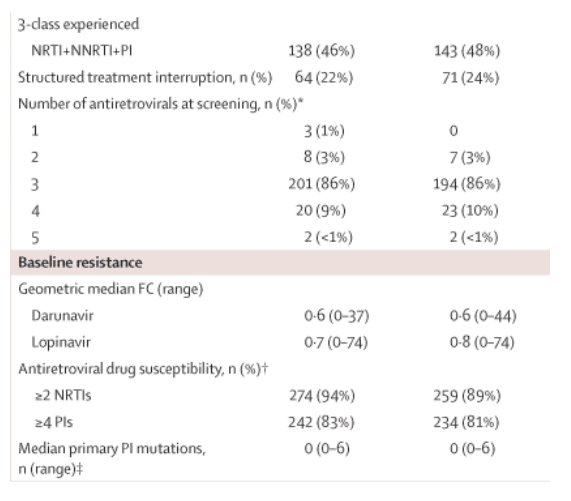
Baseline resistance characteristics were balanced between the groups, with a median phenotypic sensitivity score of underlying therapy of 2 (range 0-4) and a median of zero (0-6) IAS-USA-listed primary protease inhibitor mutations.17 Based on the phenotypic profile, 476 (82%) of patients with available data had phenotypic susceptibility to four or more protease inhibitors, and 384 (66%) were susceptible to all commercially available protease inhibitors. Overall, nine patients (2%) had darunavir FC greater than 10, of whom only one (<1%) had FC greater than 40; of the 58 patients (10%) who had lopinavir FC greater than 10, 13 (2%) had FC greater than 40. In the darunavir-ritonavir group, five patients (2%) had darunavir FC greater than 10; none had FC greater than 40. In the lopinavir-ritonavir group, of the 29 patients (10%) who had lopinavir FC greater than 10, eight (3%) had FC greater than 40.
In the per-protocol analysis, significantly more patients in the darunavir-ritonavir group than in the lopinavir-ritonavir group had confirmed plasma HIV RNA of less than 400 copies per mL at week 48 (77% [n=220] vs 68% [n=199]; estimated least square mean difference 9%, 95% CI 2-16). Since the lower limit of the 95% CI for the difference in response between treatment arms was not lower than -12%, this result established non-inferiority of darunavir-ritonavir. The results in the ITT population were consistent with those of the per-protocol population: 77% (n=228) of patients in the darunavir-ritonavir group compared with 67% (n=199) in the lopinavir-ritonavir group achieved HIV RNA of less than 400 copies per mL at week 48 (estimated least square mean difference 10%, 95% CI 2-17; p<0·0001; figure 2). Furthermore, in the ITT analysis, the lower limit of the CI (2%) did not include 0%, thereby meeting the a-priori criterion for superiority of darunavir-ritonavir over lopinavir-ritonavir (p=0·008). Of the patients who were on structured treatment interruption at screening, 47 (73%) in the darunavir-ritonavir group and 48 (68%) in the lopinavir-ritonavir group achieved HIV RNA of less than 400 copies per mL at week 48.
As with the primary efficacy endpoint, significantly more patients in the darunavir-ritonavir than in the lopinavir-ritonavir group (71% [n=211] vs 60% [n=179], ITT-TLOVR) achieved plasma HIV RNA of less than 50 copies per mL at week 48 (estimated least square mean difference 11%, 95% CI 3-19; p=0·005; figure 2). The significant difference between treatment arms in the proportion of patients with HIV RNA less than 50 copies per mL was observed as early as week 24 (estimated least square mean difference 8%, 95% CI 1-6; p=0·024) and had increased by week 48. Additionally, the proportion of patients with at least 1 log10 reduction in HIV RNA at week 48 was higher in the darunavir-ritonavir arm than the lopinavir-ritonavir arm (77% [n=230] vs 69% [n=205]; estimated difference 8%, 95% CI 1-15; p=0·028). The mean change in plasma HIV RNA from baseline at week 48 (NC=F) was -1·95 log10 copies per mL for the darunavir-ritonavir group, which was significantly greater than the -1·72 log10 copies per mL change for the lopinavir-ritonavir group (estimated difference -0·20 log10 copies per mL, 95% CI -0·39 to -0·004; p=0·046; figure 2). The median change from baseline in CD4 cell count (NC=F) at week 48 was similar between the groups: 88 cells per μL for darunavir-ritonavir and 81 cells per μL for lopinavir-ritonavir.
In each subgroup assessed, a greater number of patients in the darunavir-ritonavir group reached HIV RNA of less than 50 copies per mL than in the lopinavir-ritonavir group (ITT, figure 3, see figure 3 below). Although the study was not powered to show non-inferiority and superiority in subgroups, the results from the protocol-specified subgroup analyses (ITT) indicated that in most subgroups, the virological response (HIV RNA <50 copies per mL) in the darunavir-ritonavir group was non-inferior to that in the lopinavir-ritonavir group, and was superior in some subgroups.
In patients with baseline lopinavir FC less than or equal to 10, the darunavir-ritonavir group had more patients with HIV RNA less than 50 copies per mL (70%, 184 of 263) than did the lopinavir-ritonavir group (63%, 164 of 261). In patients with baseline lopinavir FC greater than 10, 72% (21 of 29) in the darunavir-ritonavir group compared with 28% (eight of 29) in the lopinavir-ritonavir group reached this endpoint (figure 3). A similar result was seen in patients with baseline darunavir FC 10 or lower (darunavir-ritonavir 70% [202 of 287] vs lopinavir-ritonavir 59% [170 of 286]) and darunavir FC greater than 10 (60% [three of five] vs 50% [two of four]; figure 3).
The development of resistance was studied in terms of virological failure in patients treated with darunavir-ritonavir (10%; n=31; baseline and endpoint resistance data available for n=28) or lopinavir-ritonavir (22%; n=65; baseline and endpoint genotype data available for n=56 and phenotype data for n=54). Fewer patients with virological failure in the darunavir-ritonavir than in the lopinavir-ritonavir group developed additional mutations during the treatment period: 21% (six of 28) versus 36% (20 of 56) primary protease inhibitor mutations and 14% (four of 28) versus 27% (15 of 56) NRTI resistance-associated mutations (described in detail elsewhere18). Among patients with virological failure, loss of susceptibility to protease inhibitors or NRTIs after treatment occurred in more patients in the lopinavir-ritonavir group than in the darunavir-ritonavir group; 33% (18 of 54) in the lopinavir-ritonavir group and 14% (four of 28) in the darunavir-ritonavir group were susceptible to fewer protease inhibitors (excluding darunavir) compared with baseline. Furthermore, 24% (13 of 54) in the lopinavir-ritonavir group lost susceptibility to lopinavir (FC >10) compared with 11% (three of 28) in the darunavir-ritonavir group who lost susceptibility to darunavir (FC >10). After treatment failure, 14% (four of 28) in the darunavir-ritonavir group compared with 32% (17 of 54) in the lopinavir-ritonavir group were susceptible to fewer NRTIs than at baseline, and 11% (three of 28) compared with 26% (14 of 54), had lost susceptibility to NRTIs that were used in the OBR.
The majority of adverse events (64%) were grade 1 or 2, and discontinuations due to adverse events were infrequent: 20 (7%) in the darunavir-ritonavir group and 21 (7%) in the lopinavir-ritonavir group (table 2, see table 2 below); discontinuations because of diarrhoea or because of liver and lipid abnormalities were more frequent in the lopinavir-ritonavir than in the darunavir-ritonavir group. The rate of discontinuation for skin and subcutaneous events was low: two patients in the darunavir-ritonavir group (both because of rash) and two in the lopinavir-ritonavir group (one due to lipoatrophy and another due to lipohypertrophy).
During the study, four patients in the darunavir-ritonavir group and one in the lopinavir-ritonavir group developed an AIDS-defining illness, which were reported as adverse events. Of the four patients in the darunavir-ritonavir group, one developed Kaposi's sarcoma and three developed infections (one case each of oesophageal candidosis, progressive multifocal leucoencephalopathy, and pulmonary tuberculosis). In the lopinavir-ritonavir group, the patient developed AIDS encephalopathy.
Five patients died during the treatment period (two in the darunavir-ritonavir group and three in the lopinavir-ritonavir group). The causes of death were reported as: respiratory failure secondary to Kaposi's sarcoma and septic shock caused by lobar pneumonia for the two patients in the darunavir-ritonavir group; cardiorespiratory arrest secondary to bronchopneumonia and cardiorespiratory arrest for two in the lopinavir-ritonavir group. The cause of death for the third patient in the lopinavir-ritonavir group was unknown at the time of the report. All deaths were considered to be unrelated to study treatment.
In patients on darunavir-ritonavir, the most frequently reported adverse events (incidence of 10% or greater), regardless of severity and cause, were diarrhoea (95, 32%), nausea (55, 18%), nasopharyngitis (37, 12%), headache (33, 11%) and upper respiratory tract infection (30, 10%). Among patients on lopinavir-ritonavir, diarrhoea (124, 42%), nausea (62, 21%) and nasopharyngitis (33, 11%) were most frequently reported. When adverse events were assessed according to grade 2-4 events that were at least possibly related to study treatment and occurred in 2% or more of patients, the rate of grade 2-4 diarrhoea in the lopinavir-ritonavir group was twice that in the darunavir-ritonavir group (table 2).
Rash-related events (regardless of severity or cause) were seen in 16% (n=48) of patients in the darunavir-ritonavir and 7% (n=20) in the lopinavir-ritonavir group. Two patients had a grade 3 rash-related event with darunavir-ritonavir, one of which led to permanent discontinuation and the other was reported in a patient receiving nevirapine as part of the OBR and reported to be doubtfully related to treatment. Both cases resolved without sequelae. No patient in the lopinavir-ritonavir group had grade 3 or 4 rash-related events.
Frequency of raised total cholesterol (grade 2-3, no grade 4 classification in Division of AIDS grading scale) was similar for patients in the darunavir-ritonavir (32%, n=94) and lopinavir-ritonavir groups (29%, n=86). However, fewer patients in the darunavir-ritonavir group had grade 2-4 elevation of triglycerides than in the lopinavir-ritonavir group (table 2).
At week 48, the mean absolute and percentage change in lipid concentration from baseline were similarly small for the two treatment groups. In the darunavir-ritonavir group, the differences from baseline were 0·52 mmol/L (46·5%) for triglycerides, 0·44 mmol/L (12·2%) for total cholesterol, 0·05 mmol/L (7·6%) for high-density lipoprotein, and 0·20 mmol/L (16·7%) for low-density lipoprotein. In the lopinavir-ritonavir group, the differences were 1·03 mmol/L (81·4%), 0·66 mmol/L (17·3%), 0·10 mmol/L (14·3%) and 0·20 mmol/L (14·6%), respectively. The two treatment groups had similar incidences of grade 2-4 elevations of alanine aminotransferase and aspartate aminotransferase (table 2).
Figure 3. Subgroup analysis showing proportion of patients who achieved HIV RNA <50 copies per mL (ITT)
Mean difference (95% CI) shown between two treatment groups in each subgroup. The size of each point indicates sample size assessed per group. Reference lines at -12% and 0 on y-axis indicate the lower CI threshold above which non-inferiority and superiority of darunavir-ritonavir over lopinavir-ritonavir, respectively, are established.

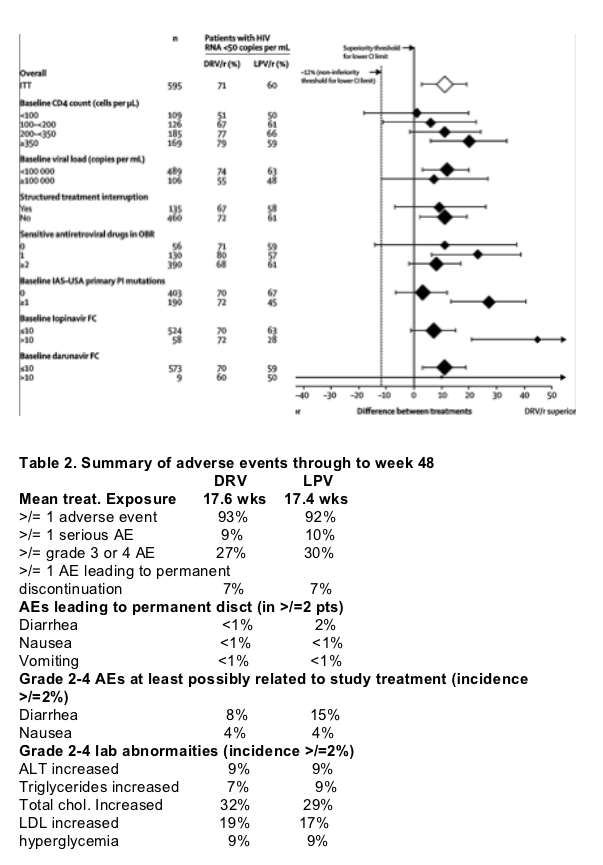
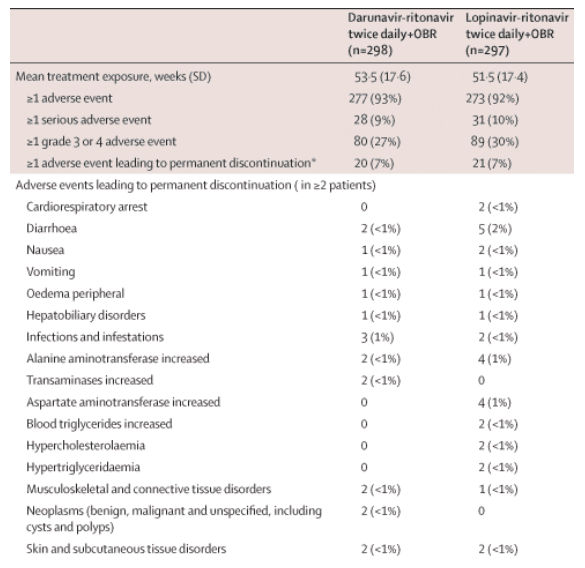
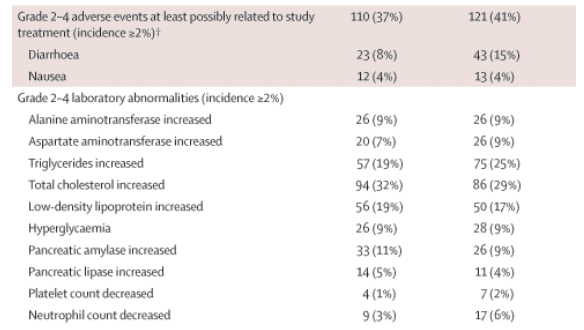
Table 1. Baseline demographics and disease characteristics
Discussion
The results of this large randomised phase III trial in lopinavir-naive, HIV-infected, treatment-experienced patients, showed that darunavir-ritonavir was non-inferior to lopinavir-ritonavir, as determined by the primary endpoint of less than 400 copies per mL of HIV RNA at week 48. Results of a secondary analysis showed that darunavir-ritonavir was superior to lopinavir-ritonavir at this timepoint. Importantly, the population was specifically selected to include patients with less advanced disease than those studied in the POWER trials, in which darunavir had already shown superiority to lopinavir and other available protease inhibitors.1,19,20 Patients in the POWER trials were allowed to use enfuvirtide, which has shown potent antiretroviral activity in heavily treatment-experienced patients, but has not been well studied in patients at earlier stages of HIV infection.12,21 To ensure that the selected patients in the present study had less advanced disease than those in the POWER studies, enfuvirtide was not allowed in the background regimen. Baseline characteristics were well distributed between the study groups, and therefore the differences observed indicate the intrinsic antiviral efficacy of the regimens studied rather than selection biases.
Our findings confirm the observation from the POWER studies that darunavir-ritonavir has a high barrier to resistance.1,19,20 In this study, darunavir-ritonavir was associated with a low rate of developing additional mutations conferring resistance to protease inhibitors and NRTIs; this can help to preserve future treatment options.
Although viral susceptibility to lopinavir was not required for eligibility, lopinavir-ritonavir would have been a reasonable clinical choice for these patients at the time, because all but eight patients in the lopinavir-ritonavir group had a lopinavir FC below the recommended upper clinical cutoff of 40. Furthermore, the benefit of darunavir-ritonavir was shown in patients who had viruses that were fully sensitive (FC <10) and in those with reduced susceptibility (FC >10) to lopinavir, showing that the results were not only driven by reduced susceptibility to lopinavir. Indeed, the lopinavir-ritonavir efficacy results seen in this study are consistent with previously reported findings in treatment-experienced patients.3,4,6,22 Therefore, the significant difference in efficacy between the regimens was unlikely to be due to suboptimal activity of lopinavir-ritonavir in this study.
These findings expand on our understanding of what can be expected from darunavir-ritonavir in various clinical settings. The results of the phase IIb POWER studies showed the antiviral and immunological benefits of darunavir-ritonavir in adults with extensive protease inhibitor resistance.1,19,20 Patients in the POWER trials had advanced disease (mean baseline HIV RNA of 4·61 log10 copies per mL and median CD4 count of 153 cells per μL) and extensive previous treatment experience (95% had used two or more protease inhibitors, and 63% were not sensitive to any commercially available active protease inhibitors). By contrast, the patients in the present study had less advanced disease, less treatment experience and were naive to lopinavir, darunavir, tipranavir, and enfuvirtide. Additionally, resistance testing showed that most patients harboured virus that was susceptible to all commercially available protease inhibitors. Therefore several therapeutic options were available for these patients at the time of enrolment.
A limitation of the study is that results of resistance testing at baseline can underestimate the degree of archived resistance (especially in patients with structured treatment interruptions). However, we controlled for this factor by stratifying patients on structured treatment interruption in each treatment group.
Immunological responses at week 48 were similar for both treatment groups, with about half the patients in each arm reaching a median CD4 count (NC=F) of greater than 350 cells per μL. This similarity, despite the difference in virological efficacy between the groups, could be explained by the fact that CD4 is a less sensitive marker of efficacy than HIV-RNA undetectability.23 The median increases in CD4 count from baseline in the darunavir-ritonavir group were consistent with those in the POWER trials at week 48.1
The types and incidences of adverse events in the darunavir-ritonavir group in this trial were consistent with those noted in the POWER studies.1,19,20 The two treatment regimens seemed to be generally equally well tolerated in this population, although discontinuations due to diarrhoea and to liver and lipid abnormalities were more frequent in the lopinavir-ritonavir group. As in the POWER studies, grade 3-4 rash occurred infrequently in the darunavir-ritonavir group.
Both protease-inhibitor-based regimens in the trial increased triglyceride concentrations. However, the mean percentage change in triglycerides from baseline was greater in patients treated with lopinavir-ritonavir than in those on darunavir-ritonavir. Moreover, the incidence of grade 2-4 hypertriglyceridaemia possibly related to treatment was 5% or greater in both groups. It is possible that ritonavir-boosting could exacerbate the effect of protease inhibitors on triglycerides, but the extent of its role has yet to be ascertained. Because of its long terminal half-life (15 h) and relative efficacy in the POWER studies at 24 weeks (38% with <50 copies per mL, 12% for control),19,20 the development programme of darunavir-ritonavir is currently studying a once-daily dose of 800 mg with 100 mg ritonavir in treatment-naive patients against lopinavir-ritonavir (ARTEMIS study). Results from ARTEMIS will improve understanding of any role of ritonavir 100 mg twice daily in raising lipid concentrations and assess whether the once-daily dosing with only 100 mg of ritonavir will lead to an improved lipid profile compared with that of lopinavir-ritonavir. Additionally, the 800/100 mg once-daily dose is being studied in treatment-experienced patients with one or fewer darunavir resistance-associated mutations.
Although new agents are expected to increase therapeutic options for highly treatment-experienced patients, the use of existing antiretroviral classes needs to be optimised to increase the effectiveness of background regimens. In view of the lower incidence of treatment failure and lower emergence of mutations seen in patients on darunavir-ritonavir, our findings show that darunavir-ritonavir provides highly effective therapy across the range of treatment-experienced patients.
Methods
Patients and study design
The TITAN trial included treatment-experienced, lopinavir-naive, HIV-1-infected patients aged at least 18 years and had a plasma HIV-1 RNA concentration of greater than 1000 copies per mL. Treatment experience was defined as being on HAART (potent anti-HIV treatment, usually including a combination of three or more drugs with activity against HIV) for at least 12 weeks. Susceptibility to lopinavir was not required for eligibility; however, all patients with previous lopinavir treatment were excluded. At the time of recruitment of this study, susceptibility cutoffs had not been defined for darunavir. In conjunction with previous treatment history, resistance testing data (Virco TYPE HIV-1, Virco BVBA, Mechelen, Belgium) for all approved antiretroviral drugs were forwarded to the investigator to assist in the selection of an OBR. Patients on a structured treatment interruption for a minimum of 4 weeks at the time of screening were also eligible. Patients co-infected with chronic hepatitis B or C were allowed to enter the trial if their condition was clinically stable and they were not expected to require hepatitis treatment during the study period. Patients with these conditions were excluded: active AIDS-defining illness (except stable Kaposi's sarcoma and wasting syndrome), any clinically important disease, and clinical or laboratory evidence of significantly decreased hepatic function at screening. Patients with previous or current use of lopinavir, darunavir, tipranavir, or enfuvirtide, or current use of investigational antiretroviral drugs, were also excluded.
TITAN is a continuing, international, randomised, controlled, open-label, 96-week phase III trial, with data obtained from patients in 159 centres in 26 countries. Patients were recruited from Apr 28, 2005, to Jan 17, 2007. The prespecified primary analysis was done when all patients had either discontinued or been treated for 48 weeks.
The study was designed in accord with guidelines of the US Food and Drug Association (FDA) and the European Agency for the Evaluation of Medicinal Products (EMEA) after discussions with regulatory authorities.7-9 Important considerations for the phase III programme of darunavir were to provide long-term controlled efficacy and safety data on the approved dose in a broad range of treatment-experienced patients. We therefore chose a well-established comparator expected to provide adequate response rates in the specified patient population. Patients were randomised in a 1:1 ratio to receive darunavir-ritonavir 600/100 mg twice daily or lopinavir-ritonavir 400/100 mg twice daily. During the baseline visit, the investigator randomised patients by calling a centralised interactive voice response system, with a manual available at each site as a guide. Randomisation was done using a predefined list constructed via random permuted blocks to ensure balance across treatment groups and across two stratification factors (use or not of a non-nucleoside reverse transcriptase inhibitor [NNRTI] in the OBR, and screening plasma HIV RNA of <50 000 copies per mL or ≥50 000 copies per mL). Darunavir was administered as 300-mg tablets, ritonavir as 100-mg capsules, and lopinavir-ritonavir as 133·3/33·3-mg capsules. During the study, a new formulation of lopinavir-ritonavir (200/50-mg tablet) became available. The study protocol was amended to allow patients randomised to lopinavir-ritonavir to switch to the new formulation as soon as it was approved by local regulatory authorities; subsequently, all patients in this arm were switched to the new formulation.
In addition to darunavir-ritonavir or lopinavir-ritonavir, patients received an investigator-selected OBR, consisting of at least two antiretroviral drugs (nucleoside reverse transcriptase inhibitors [NRTIs] with or without NNRTIs) on the basis of screening resistance data and history of antiretroviral treatment. Enfuvirtide and investigational antiretroviral drugs (except tenofovir and emtricitabine, which are still investigational in some of the participating countries) were not allowed in the OBR. For cases in which an NNRTI was included in the OBR, the dosage of lopinavir-ritonavir was increased as recommended by the prescribing information to 533/133 mg twice daily for the capsule10 and to 600/150 mg twice daily for the tablet formulation.11 The dosage of darunavir-ritonavir was 600/100 mg twice daily for all patients. No changes to the treatment regimen were allowed until the end of the treatment period, unless a drug-related adverse event occurred. Patients who had virological failure, a treatment-related grade 4 adverse event, or a confirmed grade 4 laboratory abnormality were eligible to participate in the planned rollover phase. Patients with virological failure included non-responders (plasma HIV RNA concentration never <400 copies per mL by week 16) and rebounders (initial HIV RNA <400 copies per mL followed by two consecutive tests with HIV RNA ≥400 copies per mL by week 16, or discontinuation of treatment after one test with HIV RNA ≥400 copies per mL). Since this was an open-label trial, all patients with treatment failure were reviewed, to prevent patients receiving lopinavir-ritonavir from artificially failing their treatment to gain access to darunavir-ritonavir. Virological profiles were assessed to confirm that patients fulfilled the pre-specified criteria before starting the rollover period.
Written informed consent was obtained from all patients. Study protocols were reviewed and approved by the appropriate institutional ethics committees and health authorities, and were undertaken in accordance with the Declaration of Helsinki.
Efficacy and safety assessments
Efficacy and safety variables were assessed at screening, at baseline, and at each visit (every 4 weeks until week 16, at week 24, and every 12 weeks until the end of the treatment period). The primary efficacy outcome was the proportion of patients with confirmed HIV-1 RNA of less than 400 copies per mL in plasma at week 48. Predefined secondary efficacy outcomes included: proportion of patients with plasma HIV RNA of less than 50 copies per mL (which is now seen as the virological target for all treatment-experienced patients12-14); proportion with at least 1·0 log10 reduction from baseline in plasma HIV RNA; mean change in log10 HIV RNA at all time points; and median change in concentration of CD4-positive T lymphocytes (CD4 count). Time-to-loss of virological response (TLOVR) algorithm was used to assess virological response (HIV RNA <400 and <50 copies per mL). Non-completer considered as failure (NC=F) analysis was used to assess mean change in log10 HIV RNA and immunological response.
Plasma HIV-1 RNA was measured with Roche Amplicor HIV-1 Monitor test (version 1.5; Roche Molecular Systems, Branchburg, USA). Immunological differences were assessed by absolute changes in CD4 count. Viral phenotypic and genotypic assessments were done with Antivirogram and Virco TYPE HIV-1 assays (Virco BVBA, Mechelen, Belgium), respectively. For definition of phenotypic resistance, fold change in half maximal effective concentration (FC) values of 10 and 40 were used for darunavir15 and lopinavir.10 The samples taken at screening, baseline, week 24, week 48, or withdrawal visit were analysed when received. Samples taken at other time points were analysed at the request of the Tibotec protocol virologist if viral load was greater than 1000 copies per mL. Treatment history was recorded but historical genotype was not obtained. We investigated the effects of resistance variables on virological outcome and the development of resistance.
Safety assessments were done on the basis of adverse events data, clinical laboratory tests (haematology, biochemistry, coagulation testing, and urinalysis), cardiovascular variables (vital signs and 12-lead electrocardiograph), physical examination, and anthropometric measurements. An independent data and safety monitoring board reviewed the incidence and severity of adverse events and laboratory abnormalities, which were classified according to the Division of AIDS table for grading the severity of adult and paediatric adverse events, modified by the sponsor to include more rigorous safety assessments for some variables (eg, more stringent cutoffs for laboratory variables, particularly liver function tests, and addition of QTc interval assessment).16
Statistical analysis
The primary analysis was a non-inferiority test of treatment response (HIV RNA <400 copies per mL) between the treatment groups at week 48. The per-protocol population was used to test for non-inferiority; this group consisted of all randomised patients who received study medication and adhered to the study protocol (therefore, we excluded patients who took disallowed medication for more than 1 week). Prespecified criteria stated that darunavir-ritonavir would be judged non-inferior to lopinavir-ritonavir if the lower limit of the 95% CI for the difference in virological response between the groups did not exceed -12%. This value, which was obtained by subtracting the response to lopinavir-ritonavir from that of darunavir-ritonavir, was selected on the basis of earlier observed differences between lopinavir-ritonavir and other active regimens or between other antiretroviral drugs in similar populations4,5 and FDA guidelines on HIV RNA measurements.7
On this basis, about 250 patients in each treatment arm were needed to provide adequate statistical power to ascertain whether darunavir-ritonavir was non-inferior to lopinavir-ritonavir, with a one-sided significance level of α=0·025 and 80% power to detect the difference.
If non-inferiority were established, an a-priori secondary endpoint would be to ascertain whether darunavir-ritonavir was superior to lopinavir-ritonavir or vice versa. An intention-to-treat (ITT) population was used to assess superiority as defined by current guidelines.7-9 The US FDA and EMEA allow the assessment of both non-inferiority and superiority in the same study provided that the non-inferiority comparison and choice of delta have been specified before the start of the trial (or are based on previous clinical data, or both).7-9 Darunavir-ritonavir or lopinavir-ritonavir was judged superior if the lower limit of the 95% CI for the difference in response between the two treatment arms was greater than 0%, establishing statistical significance.
Statistical analyses were adjusted on the basis of the randomisation stratification parameters. To further assess the robustness of the results, a conservative Bonferroni multiplicity correction was used to account for the fact that two objectives (non-inferiority and superiority) were tested. This correction evaluated the two-sided significance level to a nominal significance level of 2·5% (α/2) instead of 5%. Patients who discontinued prematurely had their change in HIV RNA imputed as zero (NC=F) and were considered as virological failures (TLOVR algorithm).
The development of resistance was studied in patients with virological failure. These patients were identified by use of the TLOVR non-virological failure censored algorithm, which means that for patients who discontinued for reasons other than virological failure, the HIV RNA changes or responses at time points after discontinuation were not imputed. Moreover, patients who did not have the chance to respond to treatment (discontinued before week 16) were not included in this analysis.
The safety population consisted of patients who were treated with at least one dose of study medication. The study is registered with ClinicalTrials.gov (NCT00110877)
Role of the funding source
The trial was designed and undertaken from April 28, 2005, by Tibotec Pharmaceuticals, the study sponsor and developer of darunavir. Statistical analyses were the responsibility of Tibotec Pharmaceuticals: the analysis was done by an independent statistician from a contract research organisation (SGS, Mechelen, Belgium) and was reviewed and validated by a statistician at Tibotec. The authors had full access to all the data and the corresponding author had final responsibility to submit the manuscript for publication.
|
|
| |
| |
|
|
|