 |
|
|
| |
Sensitive Genotype Testing Finds Twice As Many Mutations Compared to Standard Genotyping and Study Reports This is associated with Virologic Failure
|
| |
| |
16th Intl HIV Drug Resistance Workshop, Barbados, June 12-16, 2007
Reported by Jules Levin
"Prevalence of Low Abundance Drug Resistant Variants by Ultra-Deep Sequencing in Chronically HIV-Infected Antiretroviral (ARV) Naive Patients and the Impact on Virologic Outcomes"
BB Simen1, K Huppler Hullsiek2, RM Novak3, RD MacArthur4, JD Baxter5, C Huang1, C Lubeski1, GS Turenchalk1, MS Braverman1, B Desany1, JF simins1, JM Rothberg1, M Egholm1, and MJ Kozal.
For the Terry Beirn Community Programs for Clinical Research on AIDS (CPCRA)
1. 454 Life Sciences/Roche Diagnostics, Branford, CT, USA.
2. University of Minnesota, MN, USA.
3. University of Illinois, Chi, Il, USA.
4. wayne state University, Detroit, MI, USA.
5. Cooper University Hospital, UMDNJ-Robert Wood Johnson Medical School, Camden, NJ, USA.
6. Yale University School of Medicine, New Haven, CT, USA.
AUTHOR CONCLUSIONS:
Utlra Deep sequencing identified a significantly larger proportion of chronically infected ARV-naive patients harboring drug resistant variants than standard genotyping.
Minority drug resistant HIV variants are common in chronically HIV-infected ARV-naive patients.
For those within the NNRTI arm of FIRST, the risk of virologic failure was significantly greater for participants with variants containing reverse transcriptase (RT) resistance mutations identified by Ultra Deep sequencing at baseline than for those without RT mutations identified.
Ultra Deep sequencing is a promising new method to identify minority resistant HIV variants in patients.
You can see in the tables below that mutations were at least twice as much using the sensitive test compared to using the standard (TRUGENE) genotype test. The study authors appear to say the viral failure rate was higher if mutations were found with the sensitive test, however I have a question regarding this. What if a patient had mutations by both standard and sensitive testing, how would this compare to patients who only had mutations found by sensitive testing? Perhaps this study addresses that but its not clear to me. Still, I think this is a very important issue that deserves better attention by industry and academic researchers with followup studies to confirm these findings and discussion on how to identify such patients.
BACKGROUND
In the absence of drug selection pressure drug resistant HIV variants will often decline to undetectable levels in patients.
Current commercially available assays cannot identify low-level resistant variants (at levels <20% of the viral population).
Low abundance drug resistant HIV variants can rapidly grow under drug selection pressure and lead to treatment failure.
Although point mutation assays can identify selected mutations at low levels, there are >100 mutations associated with HIV drug resistance.
New methods are needed to detect all the possible resistance mutations that can be present in the many different viral variants in a clinical sample.
FIRST Study Baseline HIV Drug Resistance
In a FIRST substudy of 491 HIV-infected ARV-naive participants tested for resistance at baseline, the estimated prevalence of drug resistance was 10.8% for the cohort (8.8% if RT 118I mutation excluded).
Estimated prevalence of class mutations conferring resistance:
NRTIs; 7.8%
NNRTIs: 3.0%
PIs: 0.7%
Given the small number of participants with baseline resistance by standard genotyping, the impact of baseline resistance on subsequent clinical outcomes could not be assessed.
OBJECTIVES
For chronically HIV-infected ARV-naive FIRST study participants:
Determine the prevalence of low abundance drug resistant variants using Ultra Deep sequencing.
Compare the prevalence of resistance mutations in FIRST participant's plasma samples as detected by Ultra Deep and standard sequencing.
Determine the impact of baseline resistant variants detected by Ultra Deep sequencing on virologic responses.
METHODS
A subset of ARV-naive FIRST participants (n=258) had baseline drug resistance determined by both standard genotyping (TRUGENE) and Ultra Deep dequencing (minor variant detection to 1% levels).
Participants enrolled between 1999-2002 (there's probably more drug resistance present currently, particularly NNRTI by standard or sensitive testing) and had advanced disease (median HIV VL of 143,844 c/ml and CD4 of 163).
Coded FIRST baseline RNA samples were sent to 454 Life Sciences for Ultra Deep sequencing.
454 Life Sciences was given the viral load value for the sample but was blinded to the prior resistance result. RNA samples with 10,000 IU after RNA extractionb were run.
454 Life Sciences provided the Ultra Deep sequencing result to the CPCRA Statistical Center (Univ of Minnesota).
IAS-USA 2006 mutations (n=62) were evaluated for by both standard and Ultra Deep sequencing methods.
2006 Stanford HIV drug resistance database mutations (HDRM) that had an algorithm value of >/=5 (n=144) were evaluated for by both methods.
Standard and Ultra Deep sequencing methods were compared with McNemar's test for dependent proportions.
Proportional hazard models were used to compare those with and without mutations for virologic failure (HIV RNA >1000 c/ml at or after 4 months).
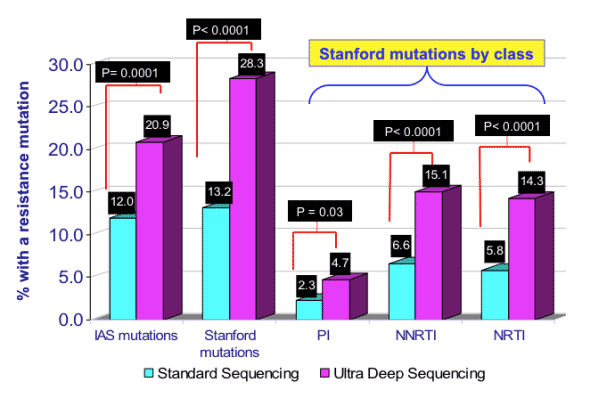
Comparison of Standard HIV Genotyping and Ultra Deep Sequencing: presence of HIV drug resistance mutations in ARV-naive chronically HIV-infected FIRST participants (n=258)
Ultra Deep sequencing found at least double the number of mutations compared to using standard genotyping.
IAS mutations: 12% using standard genotyping vs 20.9% using Ultra deep sequencing (p=0.0001).
Stanford mutations: 13.2% using standard genotyping vs 28.3% using Ultra Deep sequencing (p<0.0001).
Protease inhibitor mutations: 2.3% using standard genotyping vs 4.7% using Ultra Deep (p=0.03).
NNRTI mutations: 6.6% using standard genotyping vs 15.1% with Ultra Deep (p<0.0001).
NRTI mutations: 5.8% using standard genotyping vs 14.3% with Ultra Deep (p<0.0001).
Virologic Failure in the NNRTI Strategy
This table shows the much higher failure rate of patients with a NNRTI mutation detected by Ultra Deep sequencing Hazard Ratio 3.38 (1.35-8.43), p value 0.009 using IAS-USA mutations. Using Stanford mutations the Hazard Ratio is 3.49 (1.65-7.36) , p value <0.001). So patients were 3 times more likely to fail if they had a NNRTI mutation detected by Ultra Deep than if they Ultra deep did not detect a NNRTI mutation.

Time to Virologic Failure by Presence of NNRTI Mutations at Baseline by Ultra Deep sequencing: NNRTI strategy arm
Estimated percent with virologic failure at 12 months for those with a NNRTI mutation at baseline 72.7 vs 28.0 for those without a NNRTI mutation, p=0.006
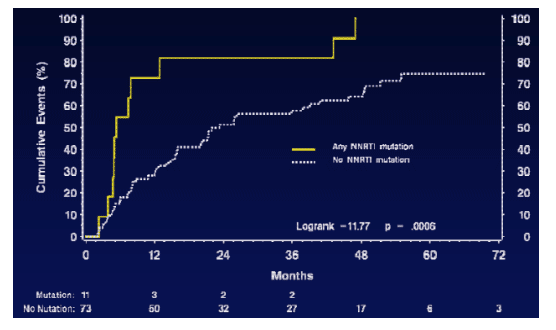
Time to Virologic Failure by Presence of RT Mutations at Baseline by Ultra Deep sequencing: NNRTI strategy arm.
Estimated percent with virologic failure at 12 months for those with a RT mutation at baseline 59.1 vs 24.8 for those without a RT mutation, p=0.004.
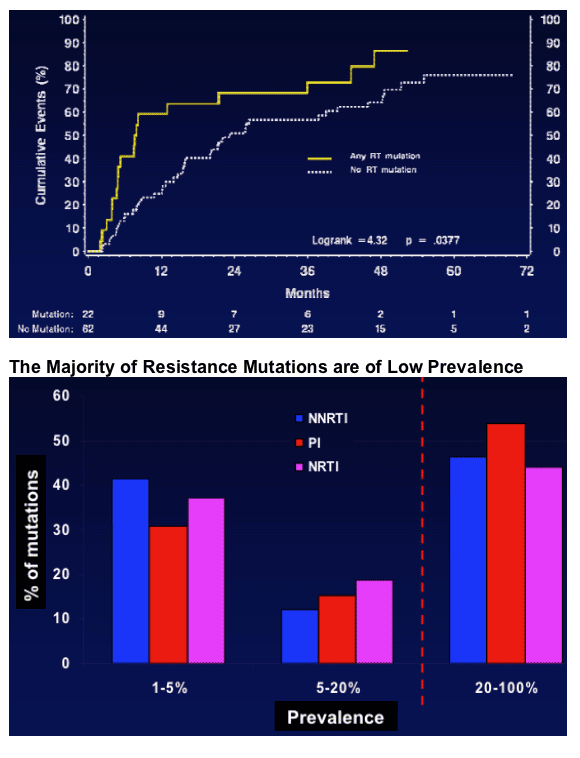
|
| |
|
|
|
|
|