 |
|
|
| |
New Roche CCR5 Potent Against Maraviroc Resistance In Vitro
|
| |
| |
Reported by Jules Levin
"CCR5 Binding Properties of a CCR5 Small Molecule Inhibitor with High Antiviral Potency Against A Maraviroc (MVC)-Resistant HIV-1 Strain"
16th Intl HIV Drug Resistance Workshop
June 12-16, 2007, Barbados
A Jekle, R Kondru, C Ji, K-T Chuang, DC Swinney, D Rotstein, S Sankuratri, N Cammack, G Heilek
Roche Palo Alto LLC, CA, USA
ABSTRACT
BACKGROUND: CCR5 inhibitors are in clinical development as promising treatment options for HIV-1 infection. We studied whether the antiviral potency of a CCR5 inhibitor against a maraviroc-resistant virus can be explained by subtle differences in the ligand-receptor interactions.
METHODS: A maraviroc-resistant virus was selected by in vitro passaging of the CCR5-tropic virus CC1/85 in peripheral blood mononuclear cells (PBMC). The resulting maraviroc-resistant virus was analysed for susceptibility to inhibitors in a PBMC infection assay. Specific binding interactions and kinetics were determined with wild-type and mutant CCR5 receptors using competitive binding methods with 3H-maraviroc as radioligand.
RESULTS: Passaging of CC1/85 in presence of maraviroc resulted in a maraviroc- and vicriviroc-resistant virus (CC1/85_MVRres). The maraviroc and vicriviroc IC50 for CC1/85_MVRres was shifted more than 8,000- and 12,000-fold compared to a control virus. In contrast, fusion inhibitors, CCR5 antibodies as well as the CCR5 small-molecule inhibitor RO1752 retained potent antiviral activity. Receptor mutation data to model receptor-ligand interactions suggest that maraviroc and RO1752 share the same binding site. However, the nature of specific interactions within the site is different. Specific interactions of antagonists within the site stabilize conformations of the receptor that are not recognized by wild-type HIV-1. The conformational changes are associated with slower binding
kinetics due to the energy barriers associated with the conformational changes. We observed that the dynamics of maraviroc binding are distinct from RO1752. Mutation of residues Y108 on helix III and Y251 on helix VI to alanine eliminated the time-dependent binding for maraviroc, but not for RO1752.
CONCLUSION: 1) A maraviroc-resistant virus was selected by in vitro passaging. 2) This virus is highly resistant to maraviroc and vicriviroc. 3) CCR5 small molecule inhibitors with good antiviral potency against the maraviroc-resistant
virus can be engineered. 4) Based on kinetic data from mutant receptor-binding-experiments, we propose that binding of inhibitors to CCR5 changes the receptor conformation. Distinct differences in the binding dynamics of individual inhibitors translate into differences in the receptor conformation. Inhibitors with antiviral potency cause conformations that can are not recognized by wildtype HIV-1. Resistant viruses learned to bind to conformations of inhibitor-bound receptors.
INTRODUCTION
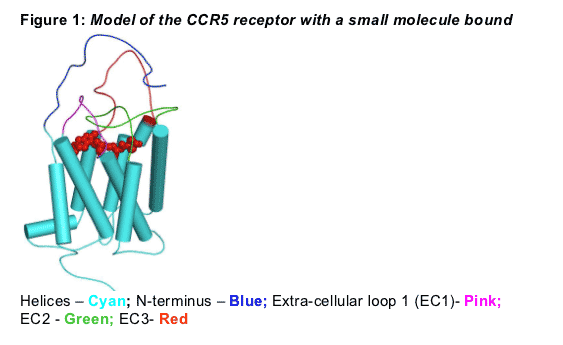
Several CCR5 inhibitors are in clinical development as promising treatment options for HIV-1 infection. All are believed to occupy a binding pocket formed at the base of the extra cellular loops of the CCR5 receptor (Figure 1). In this study we aimed to ascertain if the small molecule binding pocket for different inhibitors (e.g. MVC, RO1752) undergoes subtle conformational changes. We hypothesized that these conformational changes might explain a differential cross-resistance pattern for various small molecule CCR5 inhibitors.
AUTHOR CONCLUSIONS
Differential cross-resistance pattern can be achieved with small molecule CCR5
inhibitors.
Binding of small molecules is associated with compound specific conformational
changes in the binding site. We speculate that these conformational changes translate to compound specific changes in the extracellular loops.
Differences in binding mode of the small molecules and resulting surface changes in CCR5 can help explain the differences in the cross-resistance profile.
METHODS
In vitro passaging :
Similar to experiments reported by Westby et al.(1), the CCR5-tropic HIV-1 isolate CC1/85 was passaged in PBMC. A no-drug-control (NDC) culture was carried on in parallel (CC1/85-NDC). After 14 passages, a virus replicating at 12.5 _M MVC was isolated (CC1/85_MVRres). A tropism assay showed that both CC1/85_NDC and CC1/85_MVRres were still R5-tropic and could not be inhibited by the CXCR4 inhibitor AMD3100.
Genotypic characterization of CC1/85MVRres and CC1/85_NDC:
Full-length envelope genes were amplified by RT-PCR and cloned into the expression vector pCDNA3.1D/V5-his-TOPO (Invitrogen). Twelve clones each as well as the PCR pool were sequenced.
PBMC assay:
In a 96 well, round bottom plate, 105 PBMC were infected at a MOI of 0.001 in the presence of serially diluted drugs. Plates were incubated for 6 days at 37 C. Virus production was measured by p24 ELISA as say (PerkinElmer). IC50 was determined using a sigmoidal dose-response model with one binding site in Microsoft XLfit.
Single cycle Entry assay:
Pseudo-typed viruses were generated by transfecting 293T cells with an envdeleted pNL4-3 plasmid containing a Luciferase reporter gene and CC1/85_NDC or CC1/85_MVRres envelope expressing plasmids, respectively. Pseudovirus containing supernatant was used to infect 4x 105 PBMC in the presence of serially diluted drugs. After a three day incubation at 37 C, the Luciferase signal was determined using Ste ady-Glo (Promega).
Competition binding studies:
Non-Preincubated (NP) and Preincubated (P) IC50s were determined in parallel. P IC50s were obtained by preincubating 40_l of membrane prepared from CHO cells expressing human CCR5 receptor with 40_l of antagonist at ten half log concentrations for 3 hours, whilst for the NP IC50s, membranes were preincubated for 3 hours with buffer as a vehicle. After 3 hours 40_l of the 80_l membrane/antagonist or membrane/buffer mixes was added to reaction plates containing buffer, 3.9nM 3H -maraviroc (final concentration), and, in the case of NP IC50s, 20_l of antagonist to give a 200 _l final volume. After a 2 hour incubation period the reaction was ended using the Filtermate Harvester. The IC50s were calculated using the computer program Graphpad Prism and converted to KI using the Cheng-Prusoff correction. The KIs are reported as a mean ± SEM, of at least three separate experiments, performed in duplicate. All reactions were conducted at room temperature (22oC).
Homology Model of hCCR5:
A 3D model of the human CCR5 receptor was constructed on the basis of manual sequence alignments between bovine Rhodopsin [1] and human CCR5 sequence. Using the Protein Modeling tool within Molecular Operating Environment [2], MOE, the homology model was created from these alignments using the bovine Rhodopsin crystal structure as the backbone template for the transmembrane helices. The loop regions that are structurally close to Rhodopsin template were added to the helices by searching a library of protein structures generated from the Protein Data Bank. The side chain conformations are chosen from a rotamer library [3], and are further refined by energy minimization using CHARMM22 [4] forcefield . The final 3D structure of CCR5 receptor was obtained after a set of constrained dynamics and minimization to remove any structural constraints. A manual docking of the compounds was then carried out that was guided by mutation data.
RESULTS
Figure 2: MVC resistance mutations are clustered in the V3 loop
Mutations were identified by sequencing the entire gp160 region of five envelope clones amplified from the starting virus (WT) and 12 clones each from the MVR-res and no-drug control virus (NDC). The V3 loop is highlighted in bold, mutations compared to the WT consensus sequence in green, mutations found exclusively in CC1/85_MVRres, but not WT or CC1/85_NDC virus in red.
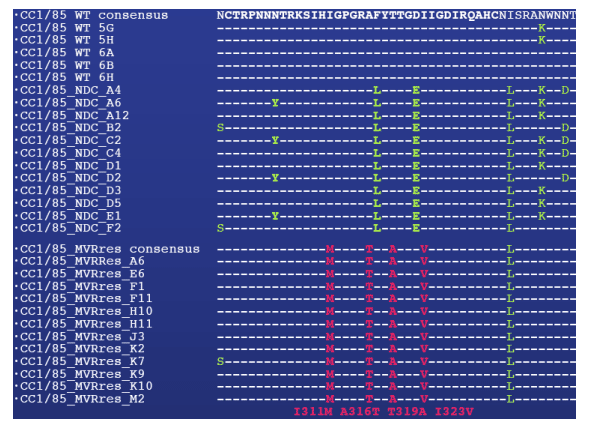
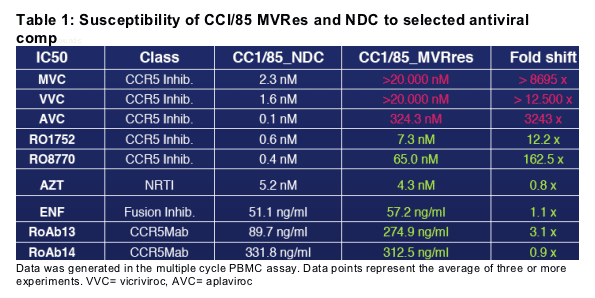
Figure 3: IC50 curves for CCI/85 MVres and NDC in A, PBMC and B, single cycle assays
CCR5 antagonists (MVC, VVC) are inactive against CC1/85_MVres in the (multiple cycle) PBMC assay ( Fig. 3a) and show reduced maximum inhibition (plateau) in the single cycle entry assay (Fig. 3b), consistent with allosteric binding models proposed by Westby et al. (5) and Pugach et al. (6).
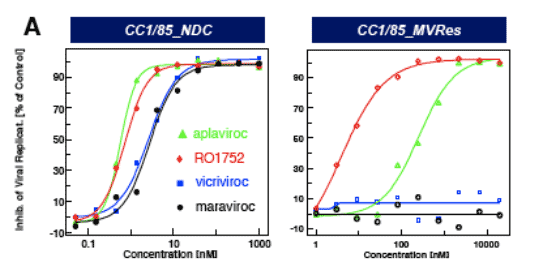
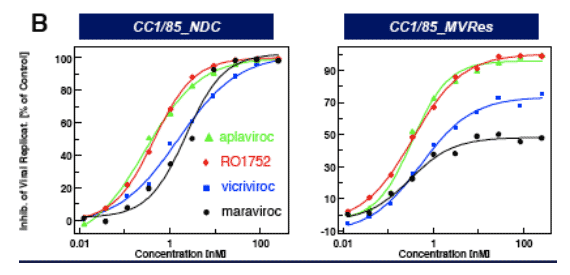
Figure 4: Model of conformational changes during CCR5 receptor-small molecule interaction
We propose the initial formation of a collision complex, followed by time dependent formation of a binding cap which includes the rearrangement of the cytosolic loops. The resulting change in the CCR5 receptor surface structure modulates the binding site for HIV gp120, preventing the viral docking. Contributions of conformational change to slow receptor binding were determined by measuring competition binding of 3H - maraviroc to the CCR5 receptor with or without 3 hr pre-incubation of unlabeled compound.
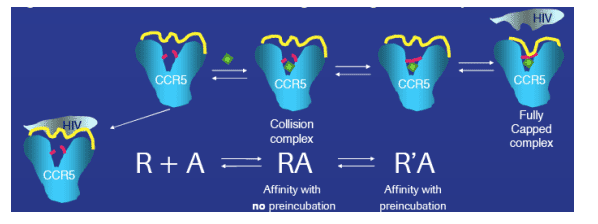
Figure 5: Sample competition binding curves
Binding and capping of the maraviroc-CCR5 complex involves residues 85, 86, 108, 198 and 251. Binding and capping of RO1752-CCR5 complex involves residues 85, 86 and 198.
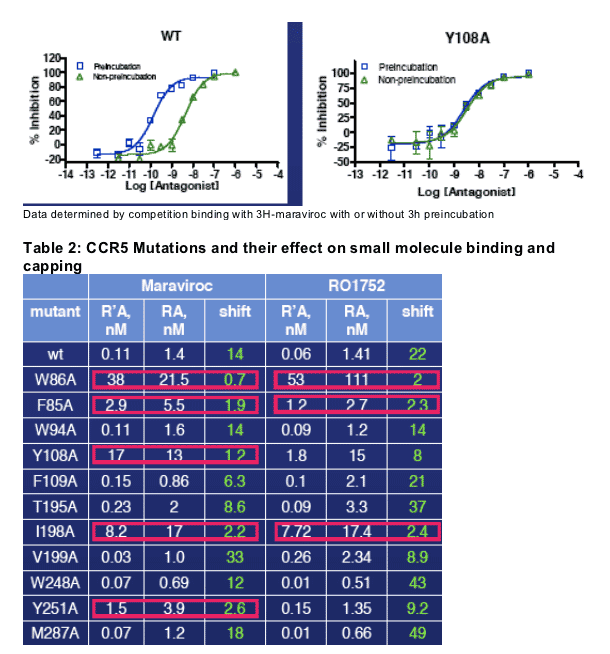
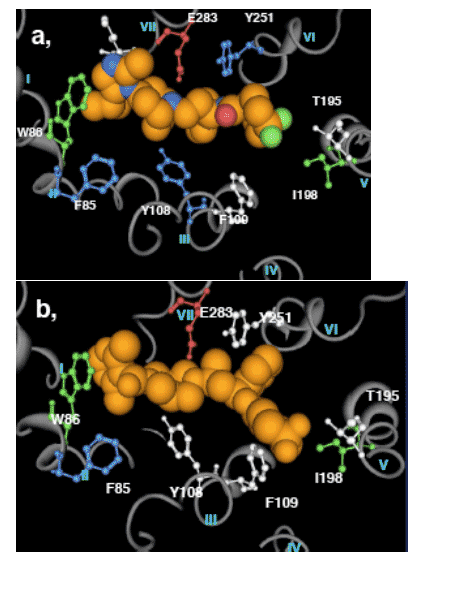
Figure 6: Dynamic binding model for a, maraviroc and b, RO1752
The conformational change to the final state is differentiated between MVC and RO1752 by the contribution of Y108 and Y251 to MVC binding. Key
residues for initial binding of both RO1752 and maraviroc are W86 and I198. F85 is involved in the conformational change to the final stable state for both compounds.
|
| |
|
|
|
|
|