| |
Tenofovir monotherapy is effective in hepatitis B patients with antiviral treatment failure to adefovir in the absence of adefovir-resistant mutations
|
| |
| |
Journal of Hepatology March 2008
Jessica Tan, Bulent Degertekin, Stephen N. Wong, Munira Husain, Kelly Oberhelman, Anna S.F. Lok
"in vitro studies show that ADV-resistant rtA181V and rtN236T mutations are susceptible to LAM and FTC [27]. Thus, combination treatment of TDF with LAM or FTC may be more effective for patients with ADV-resistance than TDF monotherapy. This concept is supported by our observation that both patients with ADV-resistance who received TDF+FTC had rapid virologic response with undetectable serum HBV DNA within 6 and 12months. Moreover, addition of FTC led to a further 1.6log decrease in serum HBV DNA in 1 patient who had ADV-resistance and suboptimal response to TDF monotherapy. Rapid viral suppression in 2 patients with ADV-resistance switched to TDF+FTC had also been reported by Villeneuve et al..... The results of our case series suggest that TDF alone may not be sufficient to suppress HBV when there are ADV-resistant mutations as these mutations persist and are further selected. Combination of TDF+FTC may be a more effective rescue therapy for patients with ADV-resistance. An alternative treatment is entecavir which has been shown to be effective in suppressing ADV-resistant HBV in in vitro studies [3] and in 2 patients [5]. Our study is limited by the small number of patients and the lack of randomization. Nevertheless, it highlights the importance of resistance mutation testing in the management of patients who have experienced sequential nucleos/tide analogue therapy and the need for studies on de novo combination therapy."
Background/Aims
We sought to identify mutations associated with treatment failure to adefovir (ADV) and to determine virologic response to tenofovir (TDF) alone and in combination with emtricitabine (FTC) in these patients.
Methods
Serum samples prior to and after the change in treatment to TDF/TDF+FTC from 13 patients were analyzed by direct sequencing and clonal analysis.
Results
ADV-resistant mutations, rtA181V and rtN236T, were detected on direct sequencing in 3 of 8 patients who had virologic breakthrough. Among patients with suboptimal virologic response, rtA181T, rtI233V, and rtN236T were present on clonal analysis in 3 patients. Ten patients received TDF, 8 achieved virologic response. One had ADV-resistance at baseline and persistence of ADV-resistant mutations during TDF treatment, addition of FTC resulted in a further decrease in HBV DNA. Another patient had no ADV-resistance at baseline, and selection of ADV-resistant mutations during TDF treatment. All 3 patients who received TDF+FTC had undetectable HBV DNA within 3-12months including 2 who had ADV-resistance at baseline.
Conclusions
TDF monotherapy is effective for patients with virologic breakthrough or suboptimal response to ADV, but combination therapy with a nucleoside analogue should be considered in patients with ADV-resistance. No novel mutations were detected.
Associate Editor: F. Zoulim
Introduction
Adefovir dipivoxil (ADV), an acyclic phosphonate, has antiviral activity against both wild type and lamivudine (LAM)-resistant hepatitis B virus (HBV). Initial studies of nucleoside-naive patients reported no evidence of drug resistance after 48weeks of ADV treatment [1]. However, ADV-resistant mutations with substitutions of valine for alanine at position 181 (rtA181V) and threonine for asparagine at position 236 (rtN236T) in the reverse transcriptase region of the HBV polymerase have been reported [2], [3]. These mutations have subsequently been observed in 29% of nucleoside-naive hepatitis B e antigen (HBeAg) negative patients after 5years of ADV treatment [4]. A higher rate of resistance has been reported in patients with LAM-resistance, who were switched to adefovir monotherapy [5], [6]. In vitro studies showed that rtA181V and rtN236T mutations decrease susceptibility to ADV by 4.3- to 23-fold [2], [3], [7]. Several other mutations have been proposed to be associated with ADV-resistance but the significance of these mutations is unclear [8], [9], [10], [11], [12]. Clinical studies have observed that as many as 50% of patients have suboptimal virologic response to ADV [5], [13]. Some possible causes for this include the low approved dose of ADV to avoid nephrotoxicity, LAM-resistance, and high HBV DNA at the start of ADV treatment. A recent report of 3 patients suggested that a novel mutation with substitution of valine for isoleucine at position 233 (rtI233V) is associated with primary non-response to ADV [14], but this finding was not confirmed in another study [15].
Tenofovir disoproxil fumarate (TDF), a nucleotide analogue closely related to ADV, has similar antiviral activity against wild type and LAM-resistant HBV as ADV in in vitro studies [16]. TDF is approved for the treatment of human immunodeficiency virus (HIV) infection; it is available on its own and as a co-formulation with emtricitabine (FTC), which has comparable antiviral activity and resistance profile as LAM. Clinical studies have observed that TDF is more effective in suppressing HBV replication than ADV [17], [18], possibly due to the higher approved dose: 300 vs. 10mg. Thus, patients with suboptimal virologic response to ADV have been reported to experience further viral suppression when treatment was switched to TDF [19]. Nevertheless, in vitro studies showed that susceptibility of HBV isolates with rtN236T and rtA181V to TDF is decreased by 4- and 3.2-fold, respectively, indicating that tenofovir may be less effective for ADV-resistant HBV [7], [20].
The aims of this study were to identify mutations associated with suboptimal virologic response to ADV and to determine virologic response to TDF, alone and in combination with FTC, in patients with suboptimal response or breakthrough during ADV treatment.
Discussion
In this study, we showed that TDF monotherapy is effective in suppressing HBV replication in most patients with virologic breakthrough or suboptimal virologic response to ADV in the absence of ADV-resistant mutations. Of the 9 patients who did not have ADV-resistant mutations on direct sequencing, 7 had rapid virologic response with undetectable serum HBV DNA within 3-15months after switching to TDF monotherapy. Our findings are in accord with those of other investigators and are likely related to the use of a higher dose of TDF vs. ADV [19], [23]. In the 2 patients who had persistent viremia, one had a 5.9log decrease in serum HBV DNA; direct sequencing and clonal analysis of the last follow-up sample did not reveal any new mutation. The other patient had only 2.4log decrease in serum HBV DNA after 27months of TDF. Clonal analysis of follow-up samples revealed the selection of mutations that had been suggested to be associated with primary non-response or resistance to ADV (rtI233V and rtA181T). However, these mutations were present in a small proportion of the clones, and are unlikely to explain the poor response to TDF.
By contrast, virologic response to TDF monotherapy was limited in the only patient who had ADV-resistant mutations on direct sequencing. After an initial 2.5log decrease, serum HBV DNA remained around 5log10copies/mL and ADV-resistant mutations persisted in the follow-up samples. Similar findings have been reported by van Bommel et al. [24].These in vivo observations confirm in vitro data that tenofovir has decreased antiviral activity against ADV-resistant HBV compared to wild type virus [25] indicating the potential for cross-resistance.
None of the patients in our study, including 4 patients who had persistent viremia after more than 12months of TDF monotherapy, was detected to have alanine to threonine substitution at position 194 (rtA194T). This mutation had been reported to be associated with TDF-resistance in one study. In that study, 2 patients with HIV/HBV coinfection receiving LAM+TDF had persistent viremia and were found to have LAM-resistant mutations and the rtA194T mutation [26]. Phenotypic studies suggested that HBV isolates with rtA194T had decreased susceptibility to TDF but this finding was not confirmed by another study [25].
In vitro studies show that ADV-resistant rtA181V and rtN236T mutations are susceptible to LAM and FTC [27]. Thus, combination treatment of TDF with LAM or FTC may be more effective for patients with ADV-resistance than TDF monotherapy. This concept is supported by our observation that both patients with ADV-resistance who received TDF+FTC had rapid virologic response with undetectable serum HBV DNA within 6 and 12months. Moreover, addition of FTC led to a further 1.6log decrease in serum HBV DNA in 1 patient who had ADV-resistance and suboptimal response to TDF monotherapy. Rapid viral suppression in 2 patients with ADV-resistance switched to TDF+FTC had also been reported by Villeneuve et al. [28].
Our study also aimed to determine if novel mutations other than rtA181V and rtN236T may contribute to virologic breakthrough or suboptimal response to ADV. Direct sequencing revealed only one mutation rtS135Y in the baseline sample of more than one patient. A previous study reported that rtY135S (reverse of what we observed) was associated with LAM-resistance [29]. However, several amino acids have been observed at this position including serine, tyrosine, and phenylalanine, suggesting that this may be a polymorphic site. Among the 4 patients with rtS135Y mutation in our study, three had virologic breakthrough but 2 of these patients also had rtA181V mutation. In the absence of phenotypic studies, the significance of rtS135Y mutation cannot be determined. The rtA181T mutation has been reported in many studies to be associated with ADV-resistance [10] although the significance of this mutation is still debated [7], [11], [25]. This mutation was not detected on direct sequencing of the baseline samples of any of our patients but was present in 1 clone in 2 patients. Among other mutations that had been suggested to be associated with ADV-resistance [8], only rtS214A was detected in 1 patient on direct sequencing and rtV84M in another patient on clonal analysis. The rtI233V mutation, which had been suggested to be associated with primary non-response to ADV, was not detected in any of our patients on direct sequencing, but was found in 1-2 clones in 3 of the 13 patients studied. In addition, this mutation was selected in the patient who had no ADV-resistant mutation on direct sequencing, and suboptimal response to TDF monotherapy. Our findings are in accord with those of Curtis et al. [15], indicating that rtI233V is not a key factor in suboptimal response to ADV. However, further studies are needed to clarify the significance of rtI233V.
The results of our case series suggest that TDF alone may not be sufficient to suppress HBV when there are ADV-resistant mutations as these mutations persist and are further selected. Combination of TDF+FTC may be a more effective rescue therapy for patients with ADV-resistance. An alternative treatment is entecavir which has been shown to be effective in suppressing ADV-resistant HBV in in vitro studies [3] and in 2 patients [5]. Our study is limited by the small number of patients and the lack of randomization. Nevertheless, it highlights the importance of resistance mutation testing in the management of patients who have experienced sequential nucleos/tide analogue therapy and the need for studies on de novo combination therapy.
Patients and methods
Patients
Adult patients with compensated chronic hepatitis B referred to the University of Michigan Liver Clinic between August 1999 and January 2007 who had suboptimal virologic response to ADV or virologic breakthrough during ADV treatment and subsequently received rescue therapy with TDF or TDF+FTC for at least 6months were included. Clinical and laboratory data were reviewed.
Quantitative HBV DNA and liver panel were tested every 3months and patients were assessed at 6- to 12-month intervals. Serial serum samples were collected before, and every 6-12months after change in therapy. All samples were stored at -80ºC. Written informed consent for the collection of blood samples was obtained from all patients and the study was approved by the Institutional Review Board at the University of Michigan.
HBV quantification
HBV DNA was quantified using COBAS Amplicor HBV Monitor Assay (Roche, Branchburg, NJ), which has a lower detection limit of 200copies/mL. Samples with values >100,000copies/mL were retested after 1:100,000 dilutions.
Nested polymerase chain reaction and direct sequencing
DNA extraction was carried out with QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA) following the manufacturer's instructions. Nested polymerase chain reaction (PCR) was performed as described previously [21]. The amplicons spanned domains A through F of the reverse transcriptase region of the HBV polymerase gene (rt1-rt280). PCR products were purified by QIAquick PCR Purification Kit (Qiagen) and directly sequenced at the DNA sequencing core facility, University of Michigan Medical Center, using the standard protocol for the ABI 3730xl DNA Analyzer (Applied Biosystems Co., Foster City, CA). The DNA sequences were aligned using Seqmanþ II and EditSeqþ software (DNASTAR Inc., Madison, WI).
Cloning
Cloning was carried out as previously described [22]. PCR-amplified HBV DNA was cloned into pGEM T Easy Vector (Promega Co., Madison, WI), and 20-31 colonies with HBV insert were selected for each sample. Recombinant plasmid DNA was purified, electrophoresed after digestion with restriction enzymes XbaI and PstI (Roche Diagnostics Co., Indianapolis, IN), and sequenced using primers SP6 or T7. The sequences of all the clones from each sample were compared using MegAlignþ software (DNASTAR Inc., Madison, WI).
Definitions
Suboptimal virologic response was arbitrarily defined as HBV DNA >4log10copies/mL after <6 months of antiviral treatment. Virologic breakthrough was defined as <1 log10copies/mL increase in HBV DNA from nadir.
Sequences of samples collected prior to TDF or TDF+FTC treatment were compared to consensus sequences of the same HBV genotype derived from GenBank database. Sequences of follow-up samples were compared to those at the start of TDF or TDF+FTC treatment. Changes in amino acid residues were classified as one of the following categories: [1] previously observed polymorphisms, [2] novel residues at polymorphic sites, and [3] conserved site mutations.
Results
Baseline characteristics of patients before TDF or TDF+FTC
Baseline characteristics of all 13 patients who met inclusion criteria are listed in Table 1. The median age was 51years (range 35-72). Eleven patients were men and 8 were Caucasians. Six patients had genotype A infection. All patients had compensated liver disease and five had cirrhosis. Nine patients had received prior LAM; of these, 8 were switched to ADV monotherapy due to LAM-resistance, the remaining patient was switched to ADV after only 1month of LAM. Four patients received ADV as de novo therapy.
All patients were receiving ADV monotherapy at the time treatment was switched. The median duration of ADV treatment was 18 (8-52)months. Eight patients (patient 1-8) had virologic breakthrough. Six patients had HBV DNA checked at 6months, four had levels >4log10copies/mL, median decrease in HBV DNA was 3.0log10copies/mL (0.6-4.4). Five patients had suboptimal response to ADV, median decrease in HBV DNA was -0.5log10copies/mL (+0.1 to -3.1) after 6months, and -0.9log10copies/mL (-0.7 to -3.8) after 12months of ADV treatment (Table 1). Eleven patients were hepatitis B e antigen (HBeAg) positive, median HBV DNA was 6.9 (4.5-10.0)log10copies/mL, and median alanine aminotransferase (ALT) was 79 (37-334)IU/mL. Ten patients were treated with TDF and the remaining 3 received TDF+FTC.
Direct sequencing and clonal analysis of baseline samples
Three of the 8 patients with virologic breakthrough had ADV-resistant HBV mutations in the baseline samples on direct sequencing. Two had a single mutation at rtA181V, the other patient had dual mutations at rtA181V and rtN236T (Table 2).
Several previously observed polymorphic site changes were detected: substitution of threonine for isoleucine at position 16 (rtI16T), leucine for phenylalanine at position 122 (rtF122L) and alanine for serine at position 223 (rtS223A). In addition, three novel substitutions at known polymorphic sites, histidine for tyrosine at position 54 (rtY54H), arginine for histidine at position 126 (rtH126R) and proline for glutamine at position 130 (rtQ130P), were observed in more than 1 patient. Mutations at conserved sites were detected in 8 patients. Most of these mutations were found in a single patient, except a substitution of tyrosine for serine at position 135 (rtS135Y) which was detected in 4 patients (Table 2). Among the proposed possible ADV-resistant mutations at rtV84M, rtS85A, rtA181T, rtV214A, rtQ215S, rtI233V, rtP237H, and rtN238T/D, only rtV214A (substitution of alanine for valine) was detected in 1 patient (patient 1) on direct sequencing. Another patient (patient 5) had changes at positions 237 (rtP237T) and 238 (rtN238H) but the amino acid substitutions were different from those previously reported.
None of the patients with suboptimal virologic response had rtA181V or rtN236T or other putative ADV-associated mutations on direct sequencing.
On clonal analysis, five additional patients had mutations that have been suggested to be associated with ADV-resistance, 1 patient had rtN236T (patient 10), 3 patients had rtA181T and/or rtI233V (patient 5, 9, and 11), and 1 patient (patient 8) had rtV84M (Table 2). Another patient (patient 2) had a single site (rtA181V) mutation on direct sequencing and mutations at two additional sites (rtI233V and rtN236T) on clonal analysis.
Virologic response to TDF and TDF+FTC
Patients with virologic breakthrough
Two patients who had ADV-resistant mutations on direct sequencing at baseline received TDF+FTC (patients 1 and 3), both had undetectable HBV DNA after 3 and 12months, respectively.
The remaining 6 patients were treated with TDF monotherapy (Table 1, Table 3, Fig. 1). HBV DNA became undetectable in 4 patients (including 1 patient who had rtI233V in 2 of 22 clones at baseline) after 3-12months. HBV DNA remained detectable after more than 12months of TDF in 2 patients (patient 2 and 8). Patient 2 had rtA181V on direct sequencing prior to TDF and additional detection of rtM204I, rtI233V, and rtN236T in one clone each on clonal analysis. HBV DNA decreased from a baseline of 7.6 to 5.1log10copies/mL after 3months and remained at that level (Fig. 1). FTC was added at month 13 and HBV DNA decreased further by 1.6log10copies/mL to 3.1log10copies/mL at the last visit, 11months after the addition of FTC. Patient 8 had rtV84M in 1 of 23 clones at baseline, HBV DNA decreased from 7.1 to 5.4log10copies/mL 6months after switching to TDF. Thereafter, HBV DNA declined very slowly, remaining at 4.3log10copies/mL after 27months of TDF treatment.
Patients with suboptimal response
Four of the 5 patients who had suboptimal virologic response to ADV were treated with TDF monotherapy (Table 3 and Fig. 2). Three patients had undetectable HBV DNA after 3, 6, and 18months of TDF, respectively. The fourth patient (patient 10) had a decrease in HBV DNA from 7.7 to 3.2log10copies/mL after 6months. However, HBV DNA remained detectable at 3.1log10copies/mL, after 14months of TDF. The only patient who received TDF+FTC (patient 9) had undetectable HBV DNA within 3months.
Biochemical and serological response to TDF or TDF+FTC
Eleven patients had ALT above the normal range prior to switching to TDF or TDF+FTC; of these, seven had ALT normalization at the last visit (Table 1). Of the 11 patients who were HBeAg positive at baseline, 2 became HBeAg negative after 1 and 6months of TDF.
Direct sequencing and clonal analysis of follow-up samples
Direct sequencing and clonal analysis of follow-up samples with detectable HBV DNA were performed to examine the evolution of ADV-resistant mutations and the emergence of TDF- or FTC-resistant mutations.
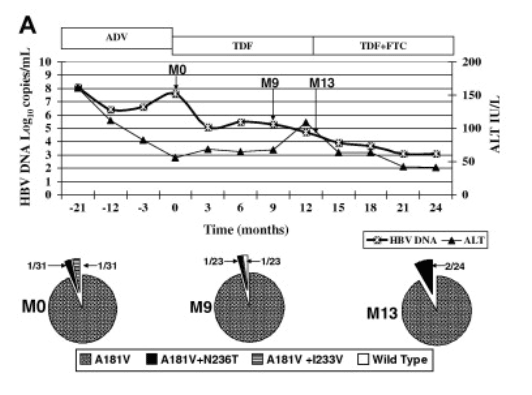
Patient 2 had rtA181V in all 31 clones at baseline, one clone had an additional rtN236T mutation and another clone had rtI233V mutation (Fig. 3A). HBV DNA decreased to 5.5log10copies/mL after 6months of TDF monotherapy and remained at that level. All 24 clones had rtA181V mutation and 2 clones had additional rtN236T mutations at month 13, when FTC was added. Blood samples after the addition of FTC were not available for testing.
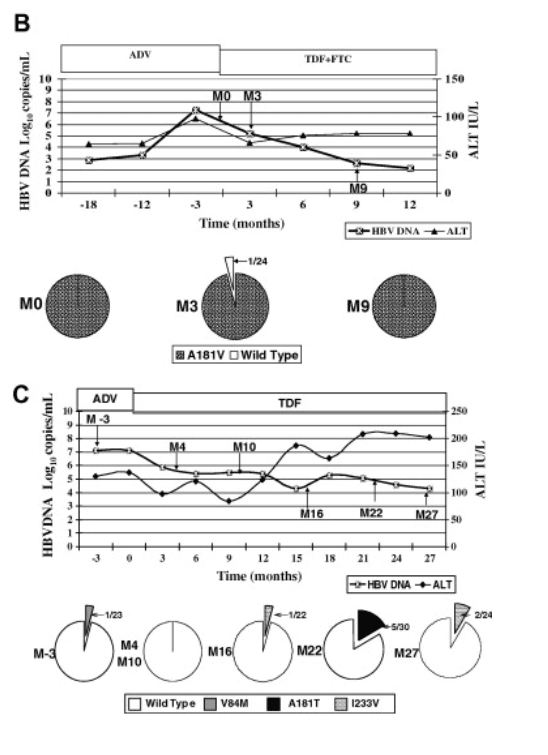
Patient 3 had rtA181V mutation in all 23 clones at baseline, HBV DNA decreased from 7.3log10copies/mL to undetectable levels after 12months of TDF+FTC treatment. At month 9, when HBV DNA had decreased to 2.6log10copies/mL, all clones still harbored the rtA181V mutation but there was no further selection of additional ADV-resistant mutations (Fig. 3B).
The third patient who had ADV-resistant mutations on direct sequencing at baseline (patient 1) had rapid virologic response to TDF+FTC and no follow-up sample with detectable HBV DNA was available for analysis.
Clonal analysis of a total of 4 follow-up samples from 3 patients (patient 5, 10, and 11), who had ADV-resistant mutations on clonal analysis at baseline, revealed wild type sequence only. Patient 9 had a rapid virologic response and HBV DNA was undetectable within 3months of treatment.
Patient 8 had suboptimal response to TDF despite the absence of ADV-resistant mutations on direct sequencing at baseline. The results of clonal analysis of five samples at different time points after the switch from ADV to TDF are shown in Fig. 3C. rtV84M, which was present in 1 of 23 clones at month 3, was not detected on clonal analysis of subsequent samples. Clonal analysis of samples at months 16, 22, and 27 revealed rtI233V and/or rtA181T mutations in 5-17% of the clones (Fig. 3C).
Alanine to threonine substitution at position 194 (rtA194T) was not detected in any of the clones derived from follow-up samples of any of the patients analyzed in this study.
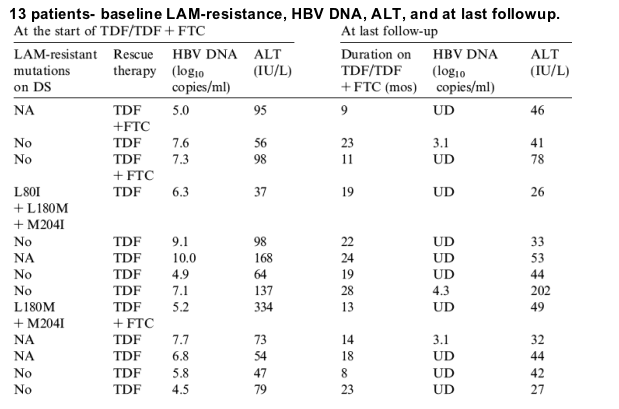
Fig. 3. (A) Clinical course and cloning results of patient 2 who had adefovir (ADV) resistance and received tenofovir (TDF) monotherapy. Pie charts showed proportion of clones with wild type and changes with established or putative association with ADV-resistance at various time points. Time 0, start of TDF. (B) Clinical course and cloning results of patient 3 who had adefovir (ADV) resistance and received combination therapy of tenofovir (TDF) and emtricitabine (FTC). Pie charts showed proportion of clones with wild type and changes with established or putative association with ADV-resistance at various time points. Time 0, start of TDF+FTC. (C) Clinical course and cloning results of patient 8 who did not have adefovir (ADV) resistant mutations on direct sequencing at baseline and suboptimal response to tenofovir (TDF) monotherapy. Pie charts showed proportion of clones with wild type and changes with established or putative association with ADV-resistance at various time points. Time 0, start of TDF.
|
|
| |
| |
|
|
|