| |
New Bone Drug Denosumab in Postmenopausal Women with Low Bone Mineral Density
|
| |
| |
NEJM Feb 23, 2006
Michael R. McClung, M.D., E. Michael Lewiecki, M.D., Stanley B. Cohen, M.D., Michael A. Bolognese, M.D., Grattan C. Woodson, M.D., Alfred H. Moffett, M.D., Munro Peacock, M.D., Paul D. Miller, M.D., Samuel N. Lederman, M.D., Charles H. Chesnut, M.D., Douglas Lain, M.D., Alan J. Kivitz, M.D., Donna L. Holloway, Ph.D., Charlie Zhang, Ph.D., Mark C. Peterson, Ph.D., Pirow J. Bekker, M.D., Ph.D., for the AMG 162 Bone Loss Study Group
ABSTRACT
Background Receptor activator of nuclear factor-B ligand (RANKL) is essential for osteoclast differentiation, activation, and survival. The fully human monoclonal antibody denosumab (formerly known as AMG 162) binds RANKL with high affinity and specificity and inhibits RANKL action.
Methods The efficacy and safety of subcutaneously administered denosumab were evaluated over a period of 12 months in 412 postmenopausal women with low bone mineral density (T score of -1.8 to -4.0 at the lumbar spine or -1.8 to -3.5 at the proximal femur). Subjects were randomly assigned to receive denosumab either every three months (at a dose of 6, 14, or 30 mg) or every six months (at a dose of 14, 60, 100, or 210 mg), open-label oral alendronate once weekly (at a dose of 70 mg), or placebo. The primary end point was the percentage change from baseline in bone mineral density at the lumbar spine at 12 months. Changes in bone turnover were assessed by measurement of serum and urine telopeptides and bone-specific alkaline phosphatase.
Results Denosumab treatment for 12 months resulted in an increase in bone mineral density at the lumbar spine of 3.0 to 6.7 percent (as compared with an increase of 4.6 percent with alendronate and a loss of 0.8 percent with placebo), at the total hip of 1.9 to 3.6 percent (as compared with an increase of 2.1 percent with alendronate and a loss of 0.6 percent with placebo), and at the distal third of the radius of 0.4 to 1.3 percent (as compared with decreases of 0.5 percent with alendronate and 2.0 percent with placebo). Near-maximal reductions in mean levels of serum C-telopeptide from baseline were evident three days after the administration of denosumab. The duration of the suppression of bone turnover appeared to be dose-dependent.
Conclusions In postmenopausal women with low bone mass, denosumab increased bone mineral density and decreased bone resorption. These preliminary data suggest that denosumab might be an effective treatment for osteoporosis.
Osteoporosis is a well-established risk factor for fracture.1 Despite treatment options that reduce the risk of fracture in patients with osteoporosis, few patients fully adhere to current therapies. A recent study reported one-year adherence rates of less than 25 percent for all osteoporosis therapies examined.2 This adherence rate is substantially lower than that for therapies for other asymptomatic conditions, such as hypertension (50 to 70 percent adherence).3 Thus, new treatment approaches that engender high adherence are needed.
Receptor activator of nuclear factor-^B ligand (RANKL), a protein expressed by osteoblastic stromal cells, binds to receptor activator of nuclear factor-B (RANK) and is the primary mediator of osteoclast differentiation, activation, and survival.4,5,6,7,8 RANKL is responsible for osteoclast-mediated bone resorption in a broad range of conditions. Osteoprotegerin, a soluble RANKL decoy receptor that binds RANKL, is the key endogenous regulator of the RANKL-RANK pathway.9
Denosumab (formerly known as AMG 162, Amgen) is a fully human monoclonal antibody (IgG2) that binds to RANKL with high affinity and specificity and blocks the interaction of RANKL with RANK, mimicking the endogenous effects of osteoprotegerin. In a phase 1 dose-escalation study, a single subcutaneous injection of denosumab resulted in a dose-dependent decrease in bone resorption, as measured by changes in serum and urinary N-telopeptide, markers of osteoclastic bone resorption.10
This article reports on a phase 2 study evaluating the efficacy and safety of denosumab in postmenopausal women with low bone mineral density.
Methods
Study Design
This randomized, placebo-controlled, dose-ranging study included eight double-blind groups and one open-label treatment group. A total of 412 women from 29 study centers in the United States were randomly assigned to receive denosumab given subcutaneously either every three months (at a dose of 6, 14, or 30 mg) or every six months (at a dose of 14, 60, 100, or 210 mg), open-label alendronate (at a dose of 70 mg) given orally once weekly, or placebo. The randomization was stratified according to center with permuted blocks of eight. Amgen prepared the randomization schedule before the study began. The denosumab solution contained denosumab (30 or 70 mg per milliliter) in 5 percent sorbitol, with 10 mM sodium acetate in water for injection (pH 5.2). All subjects took daily oral supplements containing elemental calcium (1 g) and vitamin D (400 IU). The primary end point was the percentage change in bone mineral density at the lumbar spine at 12 months. Percentage changes from baseline in bone mineral density at the total hip, femoral neck, total body (minus head), and distal third of the radius were also assessed, as were serum levels of C-telopeptide, the urinary N-telopeptide:creatinine ratio, and bone-specific alkaline phosphatase.
Institutional review boards at each study site approved the study protocol, and all subjects provided written informed consent. Amgen designed the study in collaboration with the investigators, conducted statistical analyses, and interpreted the data, which it holds. The investigators had unrestricted access to the primary data and were not limited by the sponsor in the writing of this article. The authors were responsible for writing the article and vouch for its accuracy and completeness; editing assistance was provided by Amgen.
Subjects
Postmenopausal women up to 80 years of age were eligible if they had a bone mineral density T score of -1.8 to -4.0 at the lumbar spine or -1.8 to -3.5 at either the femoral neck or total hip. An upper limit of -1.8 was selected to include subjects with both osteopenia and osteoporosis. Exclusion criteria included the use of bisphosphonates within the previous 12 months or fluoride within the previous 24 months; tibolone, parathyroid hormone or any derivative, systemic glucocorticoids (more than 5 mg of prednisone equivalent daily for more than 10 days), inhaled glucocorticoids (more than 2000 μg daily for more than 10 days), anabolic steroids or testosterone within 6 months; and estrogens, selective estrogen receptor modulators, calcitonin, or calcitriol within 3 months before enrollment. Exclusion criteria included hyperparathyroidism or hypoparathyroidism, hyperthyroidism or hypothyroidism, hypocalcemia, rheumatoid arthritis, Paget's disease of bone, osteomalacia, a creatinine clearance of less than 35 ml per minute (as estimated by the Cockroft-Gault equation),11 malabsorption syndrome, a recent long-bone fracture (within the previous six months), more than one grade 1 vertebral fracture, an osteoporosis-related fracture within the previous two years, or a case in which bone mineral density could not be accurately measured.
Study Procedures
Measurements of bone mineral density of the lumbar spine, total hip, and femoral neck were performed by dual-energy x-ray absorptiometry (GE Lunar or Hologic) at baseline and at 1, 3, 6, and 12 months and of the distal third of the radius and total body at baseline and at 6 and 12 months. Quality control and scan analysis were performed at Bio-Imaging Technologies in Newtown, Pennsylvania. Levels of serum C-telopeptide (CrossLaps, Nordic Bioscience) and urine N-telopeptide (Osteomark) in fasting samples were measured at baseline, at 3 days, and monthly through 12 months, with an additional measurement 3 days after the 6-month visit. Bone-specific alkaline phosphatase (Tandem-R Ostase, Hybritech, or Access Ostase, Beckman Coulter) and intact parathyroid hormone (Nichols) were assessed at baseline and at months 1, 3, 6, 9 (bone-specific alkaline phosphatase only), and 12.
Hematologic and chemical analyses and serum levels of denosumab and denosumab-binding antibodies were recorded at all study visits. Antibodies detected by a validated electrochemiluminescent immunoassay were screened for denosumab-neutralizing activity by a cell-based tartrate-resistant acid phosphatase bioassay. (A detailed description of the assays appears in the Supplementary Appendix, which is available with the full text of this article at www.nejm.org). Reports of adverse events were collected spontaneously and in response to nondirected questioning at each study visit.
Statistical Analysis
Summary statistical analyses of demographic and baseline characteristics were calculated for all groups. Percentage changes from baseline for measures of bone metabolism were calculated for all subjects with a baseline value and at least one value after baseline and compared across dose groups (intention-to-treat). The mean percentage changes from baseline in bone mineral density and markers of bone turnover were determined by analysis of covariance models, with treatment as the main effect and the location of the center and the baseline value of the end point as covariates. The assumption of normality was assessed with use of the Shapiro-Wilk test.12 If the assumptions were violated, a nonparametric method (van Elteren test)13 was used to corroborate the parametric results. For the primary assessment, pairwise comparisons between each group receiving denosumab and the placebo group were conducted for each efficacy measure, and the levels of significance were adjusted for multiple testing with the use of the Hochberg procedure.14 With 40 subjects per treatment group and an assumed 20 percent dropout rate, a sample size of 32 subjects per group was needed to provide 90 percent power to detect a placebo-adjusted mean difference of 3 percent at a significance level of 0.05. A common standard deviation of 3.6 percent for percentage change in bone mineral density at the lumbar spine was assumed. To facilitate dose selection for subsequent studies, exploratory comparisons between the groups receiving denosumab and the alendronate group were performed. However, these comparisons were not among the primary objectives of the study and were not prespecified in either the protocol or the statistical-analysis plan; nominal P values are thus reported. Comparisons among denosumab, alendronate, and placebo in safety analyses were likewise descriptive, with nominal P values.
Results
Subjects
Data were collected from May 2002 to April 2004. Overall, 412 subjects were enrolled and 369 (90 percent) completed 12 months of treatment. The primary reasons for early discontinuation in the denosumab, alendronate, and placebo groups were withdrawal of consent (8, 2, and 7 percent, respectively, of all randomized subjects) and adverse events (2 percent for denosumab and 2 percent for placebo, with no subjects in the alendronate group). The mean age of the study population, which was similar across groups, was 63 years (Table 1). In rounded numbers, investigators classified 85 percent of the subjects as white, 11 percent as Hispanic, and 3 percent as black. The mean baseline levels of bone mineral density and markers of bone turnover were similar across the groups.
Efficacy
Bone Mineral Density
Figure 1 and Figure 2 depict measures of bone mineral density and clinical laboratory results over time in the groups that received denosumab every three months and every six months. Denosumab treatment was associated with a mean increase in bone mineral density at the lumbar spine of 3.0 to 6.7 percent at 12 months, as compared with a decrease of 0.8 percent in the placebo group (P<0.001) (Figure 1A and Figure 2A, and Table 1 of the Supplementary Appendix). Bone mineral density at the total hip at 12 months increased by a mean of 1.9 to 3.6 percent in the denosumab groups, as compared with a decrease of 0.6 percent in the placebo group (P<0.001) (Figure 1B and Figure 2B, and Table 1 of the Supplementary Appendix). At one month, increases in bone mineral density were observed at the lumbar spine and total hip in the groups receiving denosumab (in the groups receiving 14 mg and 30 mg of drug every three months and 60 mg every six months), as compared with the group receiving placebo (P<0.05). At 12 months, the bone mineral density at the distal third of the radius increased by 0.4 to 1.3 percent in the subjects receiving denosumab, as compared with a mean loss of 2.0 percent in the placebo group (P<0.001) (Figure 1C and Figure 2C, and Table 1 of the Supplementary Appendix). At 12 months, the mean percentage change in total body bone mineral density was 0.6 to 2.8 percent in the denosumab groups, as compared with -0.2 percent in the placebo group (P<0.01, except for subjects receiving 14 mg of drug every six months) (Figure 1D and Figure 2D, and Table 1 of the Supplementary Appendix). The most effective dose of denosumab in the group receiving the drug every three months appeared to be 30 mg; for the subjects on the every-six-month regimen, 60 mg appeared optimal, since higher doses were not more effective, and a dose of 14 mg was less effective.
Alendronate increased bone mineral density, as compared with placebo, at 12 months. In exploratory comparisons, the observed mean changes in bone mineral density were at least as great with denosumab as with alendronate. These changes appeared to be greater at the distal third of the radius and total hip with denosumab at doses of 30 mg every three months and 60 mg every six months (Table 1 of the Supplementary Appendix).
Markers of Bone Turnover
The denosumab groups showed decreases in levels of serum C-telopeptide, as compared with the placebo group (P<0.001), as early as three days, the first scheduled time point after baseline (Figure 1E and Figure 2E, and Table 2 of the Supplementary Appendix). The maximum mean percentage reduction in levels of serum C-telopeptide was 88 percent among the denosumab groups, as compared with 6 percent in the placebo group. The duration of the decrease was dose-dependent. Partial reversibility in levels of serum C-telopeptide was observed in subjects receiving 6-mg doses every three months and 14-mg doses every six months; the effect was more sustained in the subjects receiving 14-mg and 30-mg doses every three months and 60-mg, 100-mg, and 210-mg doses every six months. The results of tests of the urinary N-telopeptide:creatinine ratio were similar to those for serum C-telopeptide (Table 3 of the Supplementary Appendix). There was a one-month delay in the decrease in bone-specific alkaline phosphatase levels, with reductions relative to placebo thereafter in all subjects receiving denosumab (P<0.001) (Figure 1F and Figure 2F, and Table 4 of the Supplementary Appendix).
Alendronate also reduced markers of bone turnover, as compared with placebo, throughout follow-up. Levels of serum C-telopeptide and urinary N-telopeptide decreased more rapidly, and at least as effectively, in subjects who received denosumab as in subjects who received alendronate, according to measurements taken at day 3 through month 3. Greater reductions continued to be observed through 12 months among subjects receiving the highest denosumab doses (30 mg every 3 months and 100 mg or more every 6 months) (Tables 2 and 3 of the Supplementary Appendix). Changes in bone-specific alkaline phosphatase were similar between the groups receiving denosumab and alendronate (Table 4 of the Supplementary Appendix).
Biochemical Analysis
The mean albumin-adjusted serum levels of calcium in denosumab-treated subjects demonstrated early but small decreases from baseline, as compared with the placebo and alendronate groups (Figure 1G and Figure 2G). Dose dependency of these changes was not observed. The lowest mean serum level of calcium (2.32 mmol per liter [reference range, 2.10 to 2.58]) was observed three days after the administration of 30 mg of denosumab every three months. The mean serum levels of calcium at day 3 in the alendronate and placebo groups were 2.41 and 2.44 mmol per liter, respectively. Six subjects receiving denosumab (1.9 percent) had albumin-adjusted serum calcium levels that fell below the reference range. The lowest recorded value (1.95 mmol per liter) occurred at two months in a subject who received 14 mg of denosumab every six months. These decreased values neither persisted nor were symptomatic.
Concentrations of intact parathyroid hormone increased in the denosumab and alendronate groups, as compared with placebo (Figure 1H and Figure 2H). The maximum mean level of intact parathyroid hormone in any denosumab group was 10.5 pmol per liter (reference range, 1.1 to 6.9) at one month in the group that received 210 mg of denosumab every six months (5.7 pmol per liter at baseline). For comparison, the mean concentrations of intact parathyroid hormone at one month in the placebo and alendronate groups were 5.0 and 7.8 pmol per liter, respectively. The increased levels returned toward baseline levels over time. At 12 months, the mean concentrations of intact parathyroid hormone were 5.4 and 6.5 pmol per liter in the groups that received 60 mg and 210 mg of denosumab, respectively, every six months.
Adverse Events and Safety
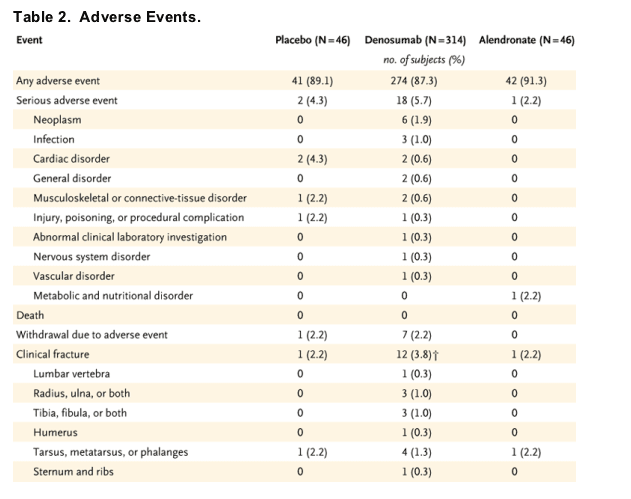
No significant differences were observed between the profiles of adverse events in the denosumab groups and those in the placebo group and the alendronate group (P>0.05), with the exception of the incidence of dyspepsia, which appeared to be significantly greater in the alendronate group (Table 2, which shows overall adverse events, and Table 3, which shows the incidence of adverse events occurring in at least 10 percent of subjects). Other than changes in levels of calcium and parathyroid hormone, as previously described, no clinically relevant changes in either blood chemistry or hematologic analysis were observed among treatment groups; there were no notable differences in group mean values, frequency of changes in toxicity grades, or individual trends over time.
Two subjects who received denosumab at a dose of 100 mg every 6 months had detectable denosumab-binding antibodies, one at 1 month and the other at 12 months. The antibodies were not neutralizing on the basis of results from a bioassay. Changes in bone mineral density and markers of bone turnover in these two subjects were within the range of responses of other subjects in their treatment group. Neither of these subjects withdrew from the study, and antibodies were not detected in subsequent samples in either subject.
Discussion
The discovery of the RANKL-RANK pathway as the primary mediator of osteoclast differentiation, activation, and survival4,7,8,9 facilitated the design of molecules that specifically target this pathway for the treatment of osteoporosis. By mimicking the effect of endogenous osteoprotegerin, denosumab, a fully human monoclonal antibody to RANKL, inhibited bone resorption with a rapid onset of action and a sustained but reversible effect.10
The present study in 412 postmenopausal women with low bone mineral density demonstrated that denosumab, given subcutaneously at three-month or six-month intervals, can increase bone density, as compared with placebo, at sites rich in trabecular bone (lumbar spine) and cortical bone (femoral neck, total hip, distal third of the radius, and total body). Formal statistical analysis for dose dependency was not prespecified and was not conducted. However, on the basis of results of the statistical analysis comparing each denosumab group with placebo, descriptive statistics, and visual inspection of the data, it appeared that doses of 30 mg of denosumab every three months and 60 mg every six months provided maximal biologic effect at the minimum exposure dose.
The increased bone mineral density at the distal radius, which is composed mainly of cortical bone, differentiated the response to denosumab from the response to alendronate at this site. This effect of denosumab on cortical bone has been observed in preclinical studies, although the mechanism remains unclear to date.
The data indicate that denosumab has a rapid onset of action. The decrease in serum levels of C-telopeptide was near maximal three days after dosing. Differences between denosumab and alendronate in changes in markers of bone resorption appeared to be more pronounced than were differences in changes of bone-specific alkaline phosphatase or bone mineral density. The latter changes are possibly less dynamic than are markers of bone resorption.
Denosumab has a long plasma-circulating time after a single subcutaneous injection.10 In this study, decreased bone turnover was sustained for approximately six months or more after single denosumab doses of 60 mg or more. This effect was reversible, as indicated by a return of serum levels of C-telopeptide toward baseline by the end of the six-month period at lower doses.
Limitations of this study include the fact that alendronate treatment was not blinded, which may have confounded comparisons of tolerability. However, bone density scans were analyzed in a blinded fashion, and the effect of alendronate on bone mineral density (a 4.6 percent increase at the lumbar spine) was similar to results of a controlled study using the same dose.15 The small number of subjects in each group precluded conclusive assessment of the effect of therapy on the incidence of fracture.
In conclusion, denosumab that was administered subcutaneously at 3-month or 6-month intervals over a period of 12 months resulted in a sustained decrease in bone turnover and a rapid increase in bone mineral density. Increases in bone mineral density observed with denosumab appeared to be superior to those with placebo and similar to or greater than those with open-label alendronate (although this study was not designed to test equivalence). These results support the continued investigation of denosumab for use in the treatment and prevention of osteoporosis and other diseases associated with bone loss.
Supported by Amgen.
Dr. McClung reports having served as a consultant to Amgen, Eli Lilly, Merck, Novartis, NPS Pharmaceuticals, Procter & Gamble, Roche, Sanofi-Aventis, and Wyeth and having received grant support from Amgen, Eli Lilly, Merck, Novartis, Organon, Pfizer, Roche, and Sanofi-Aventis. Dr. Lewiecki reports having served as a consultant to Merck, Procter & Gamble, and Eli Lilly; having received lecture fees from Procter & Gamble; and having received grant support from Amgen. Dr. Cohen reports having served as a consultant to Amgen, Abbott, and Genentech; having equity interests in Merck and Pfizer; and having received lecture fees from Abbott, Genentech, and Amgen. Dr. Cohen is medical director of Radiant Research, Dallas, which receives grant support for clinical trials. Dr. Bolognese reports having received lecture fees from Eli Lilly, Roche, and Aventis. Dr. Woodson reports having received grant support from Amgen. Dr. Peacock reports having received consulting fees from Amgen. Dr. Miller reports having served as a consultant to Merck, Eli Lilly, Wyeth-Ayerst, Roche, Procter & Gamble, and Aventis; having received lecture fees from Eli Lilly, Procter & Gamble, Aventis, Roche, Amgen, Merck, and Novartis; and having received grant support from Merck, Procter & Gamble, Aventis, Eli Lilly, Roche, Novartis, and Amgen. Dr. Lederman reports having received lecture fees from Eli Lilly. Dr. Chesnut reports having received grant support from Amgen. Dr. Lain reports having equity interests in Merck and Pfizer. Dr. Kivitz reports having an equity interest in Amgen. Drs. Holloway, Zhang, Peterson, and Bekker report having equity interests in Amgen; at the time the study was conducted, they were employees of Amgen. No other potential conflict of interest relevant to this article was reported.
We are indebted to Clifton Chunn and Holly Brenza Zoog of Amgen for their assistance in the preparation of the manuscript.
* Other members of the AMG 162 (Denosumab) Bone Loss Study Group are listed in the Appendix.
Source Information
From Providence Portland Medical Center, Portland, Oreg. (M.R.M.); New Mexico Clinical Research and Osteoporosis Center, Albuquerque (E.M.L.); Radiant Research, Dallas (S.B.C.); Bethesda Health Research Center, Bethesda, Md. (M.A.B.); Atlanta Research Center, Decatur, Ga. (G.C.W.); OB-GYN Associates of Mid Florida, Leesburg, Fla. (A.H.M.); Indiana University School of Medicine, Indianapolis (M.P.); Colorado Center for Bone Research, Lakewood (P.D.M.); Radiant Research, Lake Worth, Fla. (S.N.L.); University of Washington Medical Center, Seattle(C.H.C.); Arthritis Associates and Osteoporosis Center of Colorado Springs, Colorado Springs, Colo. (D.L.); Altoona Center for Clinical Research, Duncansville, Pa. (A.J.K.); and Amgen, Thousand Oaks, Calif. (D.L.H., C.Z., M.C.P., P.J.B.).
|
|
| |
| |
|
|
|