 |
|
|
| |
"Deep" Sequencing to Quantify in vivo Viral Fitness after Transmission of Resistant HIV
|
| |
| |
Reported by Jules Levin
ICAAC Sept 11-15 2009 San Francisco
Luke C. Swenson1, Guinevere Q. Lee1, Theresa Mo1, Conan Woods1, P. Richard Harrigan1
1BC Centre for Excellence in HIV/AIDS, Vancouver, Canada
AUTHOR CONCLUSIONS
'Deep' sequencing could detect transmitted drug resistance mutations (e.g., D30N, V108I, G190E) not detected by standard sequencing.
The rate of reversion of some transmitted resistance mutations, L10I, M46V and L90M could also be estimated from the proportion of sequences containing any mutation within the 'deep' sequencing results
Background
'Deep' sequencing, using the Roche/454 Genome Sequencer FLX (GS-FLX) is a highly sensitive technique that can detect and quantify the drug resistance profile of longitudinal samples
Drug resistance mutations present in the donor's virus tend to revert to the more fit wild-type codons when not exposed to drug selection pressure
Since the GS-FLX may be used to determine the prevalence of drug resistance mutations within a viral population, it may also be used to determine the rate of reversion (and hence fitness) of these mutations in vivo
Methods
Standard drug resistance testing (using population-based sequencing) indicated transmitted drug resistance in this patient
Plasma samples (N=8) were taken from a period of >2 years while the patient remained off therapy
The protease and reverse transcriptase (RT) regions were amplified independently in triplicate and combined in equal proportions before undergoing 'deep' sequencing with a GS-FLX
A complete protease gene and partial RT gene (codons 35-120 & 154-225) were sequenced in both the forward and reverse directions, generating a mean of >3500 sequences per gene, per direction. Of these, a mean of ∼1150 complete, in-frame, usable sequences were used to calculate the prevalence of resistance mutations.
RESULTS
A number of resistance mutations were detected at the first available timepoint, and generally reverted to wild-type over the course of the ∼2 years prior to the patient first initiating antiretroviral therapy.
Some mutations were detected by "deep" sequencing at low levels where standard, population-based sequencing indicated wild-type only
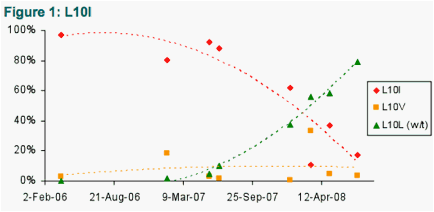
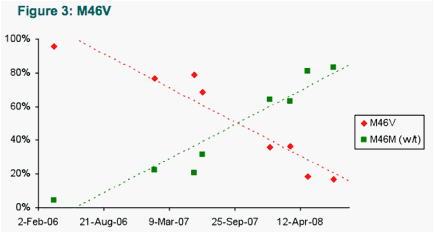
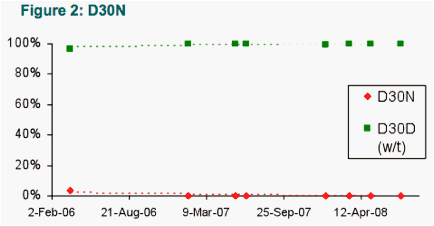
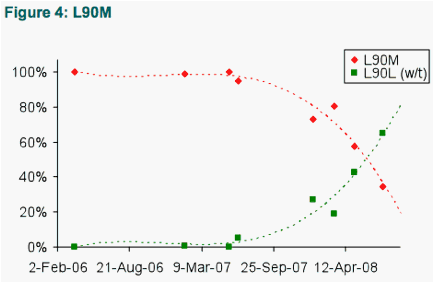
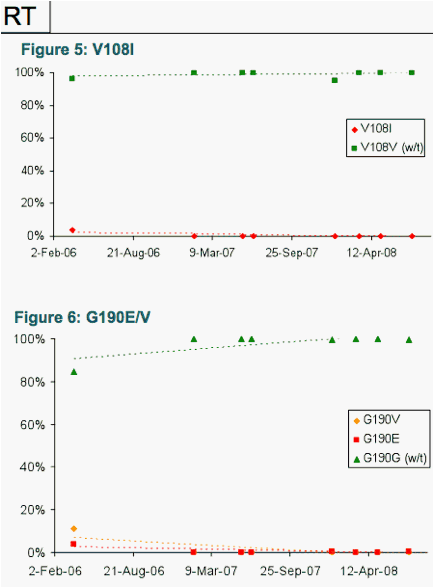
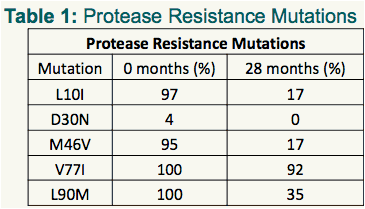
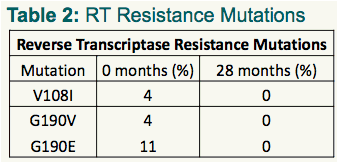
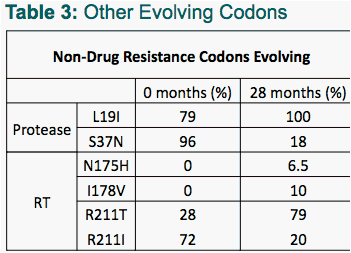
The drug-resistance mutations detected in protease and RT are shown in Tables 1 and 2, respectively. The prevalence (according to 'deep' sequencing) of a selection of these are displayed in more detail in Figures 1-4 (Protease), and 5-6 (RT), respectively.
Other evolving codons are shown in Table 3. The protease mutation N37S seemed to be transmitted to the patient, and is a known immune escape mutation associated with HLA-B44. This reverted to >80% wild-type over the study period, presumably in the absence of the HLA selection pressure.
Of the drug resistance mutations, L10I and M46V and L90M all reverted from near 0% wild-type to greater than ∼70% wild-type by the end of the study period. The rate of reversion differed between these mutations, however, with L10I and L90M exhibiting a trend of decay, but with M46V reverting more rapidly.
- The overall mean rate of change for L10I was 0.65%/week, half-time ∼22 months.
- The overall mean rate of change for L90M was ∼0.5%/week (half-time ∼ 24 months), but reverted very slowly (remaining at/near 100% prevalence) for the first ∼14 months.
- The overall mean rate of change for M46V was ∼0.64%/week (95.4% at 1st timepoint to 16.8% at final timepoint; half-time ∼18 months).
- Some mutations found at very low levels (<5%) in the first timepoint had effectively fully reverted by the second time-point (e.g., D30N in protease and V108I in RT), suggesting low in vivo fitness in the absence of drug treatment, and rapid reversion upon their transmission

|
| |
|
|
|
|
|