 |
|
|
| |
Phenotypic Screening for HIV Tropism versus both Population-based and "Deep" Sequencing
|
| |
| |
Reported by Jules Levin
ICAAC Sept 11-5 2009 San Francisco
Luke C. Swenson1, Rachel A. McGovern1, Winnie Dong1, Theresa Mo1, Guinevere Q. Lee1, Conan Woods1, Xiaoyin Zhong1, P. Richard Harrigan1
1BC Centre for Excellence in HIV/AIDS, Vancouver, Canada
Supported by Investigator Initiated Research by Pfizer
AUTHOR CONCLUSIONS
All three approaches to determining tropism give concordant results in most (>85%) cases
Discordant results were driven by factors including the detection of minority X4 variants or CD4 counts
These data may help to explain the recent success of genotypic testing for predicting clinical response to maraviroc in the MOTIVATE trials
BACKGROUND
Tropism testing is required prior to treatment with CCR5-antagonist medication such as maraviroc
Currently, the recombinant, phenotypic Trofile assay (Monogram Biosciences) is the most commonly used method to test for HIV tropism/coreceptor-usage
Genotypic testing using either standard, population-based sequencing or novel 'deep' 454 sequencing of the V3 loop of HIV env may be alternatives.
Population-based sequencing has recently been shown to be comparable to Trofile in predicting clinical response to maraviroc in the MOTIVATE trials (Harrigan et al. IAS 2009, Cape Town, Abstract WELBA101).
Here we compare all three approaches on a large number of screening samples from the MOTIVATE 1 & 2 studies of maraviroc in treatment experienced individuals, where patients were originally screened using the standard Trofile assay.
METHODS
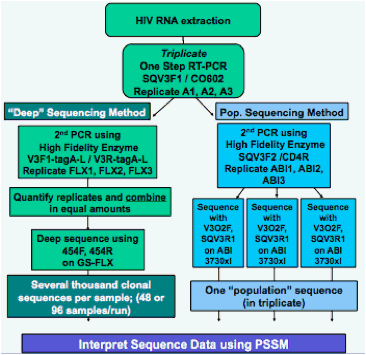
Two genotypic methods for inferring tropism were tested on screening samples from the MOTIVATE trials of maraviroc: one using population-based sequencing, and one using 'deep' sequencing (Fig. 1).
Both genotypic methods inferred tropism using the PSSM algorithm with a -2.96 cut-off. Samples analysed with 'deep' sequencing were considered non-R5 if ≥5% of their sequences were inferred as CXCR4-using. Trofile Dual/Mixed (DM) and X4 results were classified as non-R5.
Figure 1: Outline of Genotypic Methods
RESULTS
To date, a total of 1433 samples had matching results from all 3 assays.
Population-based (pop'n.) sequencing and 'deep' sequencing were concordant with Trofile in 86% and 87% of cases, respectively - comparable to their concordance with each other (85%) (Figs. 2-7).
The median %X4 by "deep" sequencing was low (0.0%) for Trofile-R5 samples, intermediate (10.8%) for Trofile-DM samples, and high (99.9%) for Trofile-X4 samples (Fig. 4).
Where Trofile and pop'n. sequencing were concordant, the median "deep" sequencing %X4 was 0% (Trofile-R5/Pop-R5) and 46% (DM/X4). Where discordant, median %X4 was low in both cases: 1.8% (R5/X4), and 1.0% (DM/R5) (Fig. 5).
Trofile-DM samples called R5 by pop'n. sequencing had X4 variants comprising <30% of their 'deep' sequencing results, suggesting that the lower reliability of pop'n sequencing in detecting minority CXCR4 using quasispecies may be driving this discordance. (Fig. 4)
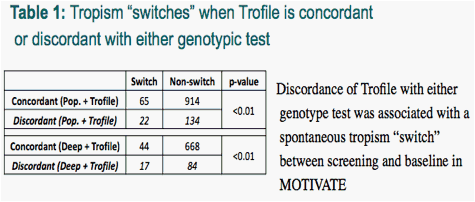
A total of 1135 samples had matching screening and baseline Trofile results. Discordance of Trofile with either genotype test was associated with the spontaneous change in the Trofile result (i.e., a tropism "switch") between screening and baseline sampling, with ∼6-7% of concordants versus ∼14-17% of discordants switching tropism (p<0.01; Chi-square test) (Table 1).
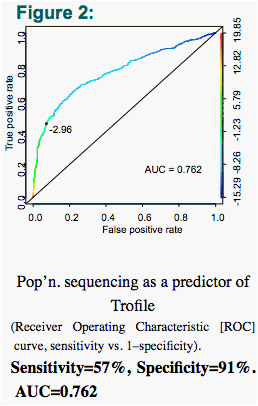
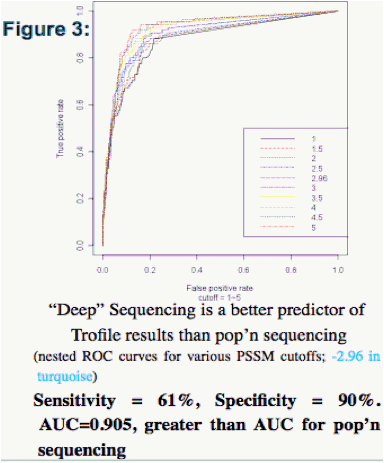
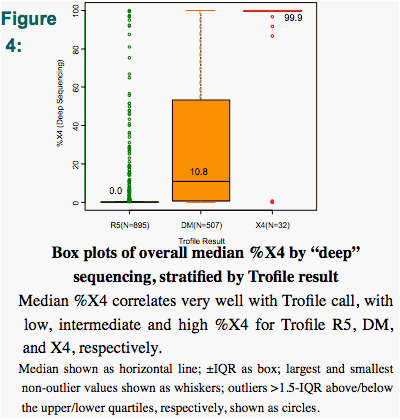
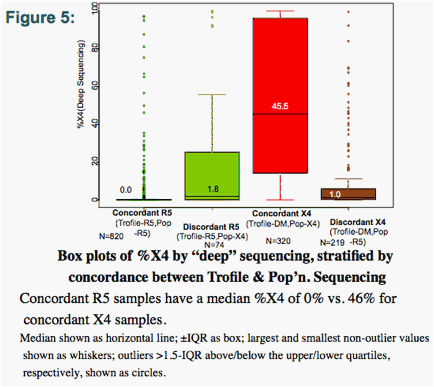
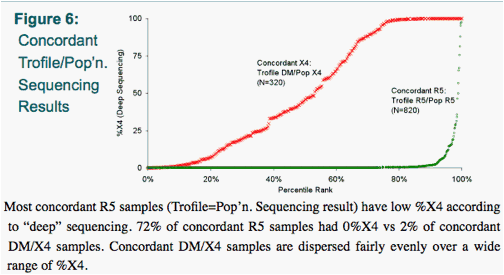
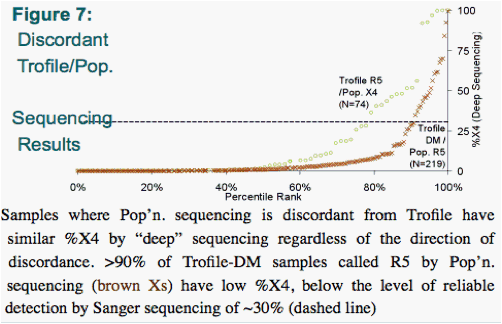
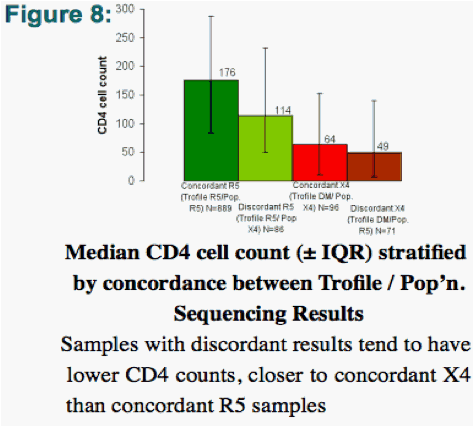
Figure 9: "Deep" sequencing results stratified by:
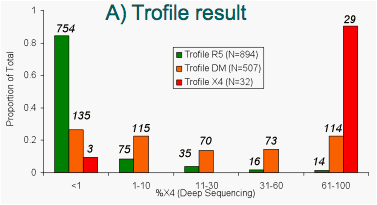
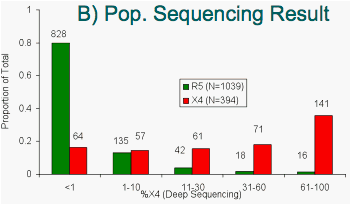
Samples called R5 by Trofile or Pop'n. Sequencing have low %X4 according to "deep" sequencing. Samples called non-R5 by either method have higher %X4. Ns shown in italics.
|
| |
|
|
|
|
|