| |
Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial
|
| |
| |
Download the PDF here
AASLD: Once Daily Sofosbuvir (GS-7977) plus PEG/RBV In Treatment-Naïve Patients With HCV Genotype 1, 4, and 6 Infection: The ATOMIC Study
http://www.natap.org/2012/AASLD/AASLD_21.htm
The Lancet, Early Online Publication, 15 March 2013
Summary
Background
The uridine nucleotide analogue sofosbuvir is a selective inhibitor of hepatitis C virus (HCV) NS5B polymerase. We assessed the safety and efficacy of sofosbuvir in combination with pegylated interferon alfa-2a (peginterferon) and ribavirin in non-cirrhotic treatment-naive, patients with HCV.
Methods
For this open-label, randomised phase 2 trial, we recruited patients from 42 centres in the USA and Puerto Rico between March 23, 2011, and Sept 21, 2011. Patients were eligible for inclusion if they had chronic HCV infection (genotypes 1, 4, 5, or 6), were aged 18 years or older, and had not previously received treatment for HCV infection. Using a computer-generated randomisation sequence, we randomly assigned patients with HCV genotype-1 to one of three cohorts (A, B, and C; in a 1:2:3 ratio), with randomisation stratified by IL28B (CC vs non-CC allele) and HCV RNA (<800 000 IU/mL vs ≥800 000 IU/mL). Patients received sofosbuvir 400 mg plus peginterferon and ribavirin for 12 weeks (cohort A) or for 24 weeks (cohort B), or 12 weeks of sofosbuvir plus peginterferon and ribavirin followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin (cohort C). We enrolled patients with all other eligible genotypes in cohort B. The primary efficacy endpoint was sustained virological response at post-treatment week 24 (SVR24) by intention-to-treat analysis. This trial is registered with ClinicalTrials.gov, number NCT01329978.
Results
We enrolled 316 patients with HCV genotype-1: 52 to cohort A, 109 to cohort B, and 155 to cohort C. We assigned 11 patients with HCV genotype-4 and five patients with genotype-6 to cohort B (we detected no patients with genotype 5). In patients with HCVgenotype-1, SVR24 was achieved by 46 patients (89%, 95% CI 77-96) in cohort A, 97 patients (89%, 82-94) in cohort B, and by 135 (87%, 81-92) in cohort C. We detected no difference in the proportion of patients achieving SVR24 in cohort A compared with cohort B (p=0·94), or in cohort C (p=0·78). Nine (82%) of 11 patients with genotype-4 and all five with genotype-6 achieved SVR24. Seven patients, all with genotype-1 infection, relapsed after completion of assigned treatment. The most common adverse events that led to the discontinuation of any study drug-anaemia and neutropenia-were associated with peginterferon and ribavirin treatment. Three (6%) patients in cohort A, 18 (14%) patients in cohort B, and three (2%) patients in cohort C discontinued treatment because of an adverse event.
and no patients who had relapse showed the presence of the signature Ser282Thr mutation in population sequencing.
Interpretation
Our findings suggest that sofosbuvir is well tolerated and that there is no additional benefit of extending treatment beyond 12 weeks, but these finding will have to be substantiated in phase 3 trials. These results lend support to the further assessment of a 12 week sofosbuvir regimen in a broader population of patients with chronic HCV genotype-1 infection, including those with cirrhosis.
Funding
Gilead Sciences.
Introduction
For previously untreated patients with chronic hepatitis C virus (HCV) genotype-1 infection, the standard of care is one of two HCV protease inhibitors-telaprevir or boceprevir-in combination with pegylated interferon alfa-2a (peginterferon) and ribavirin for up to 48 weeks.1 Duration of treatment is defined by patients' on-treatment response; the dosing schedules for both drugs allow the shortening of treatment duration to 24-28 weeks in patients with no liver cirrhosis who achieve and maintain undetectable HCV RNA in the first 8 weeks of treatment.2, 3 The potential to shorten duration of treatment is important because it can reduce the occurrence of the serious side-effects associated with peginterferon and ribavirin (headache, fever, cytopenia, autoimmunity disorders, and depression).4, 5 Unfortunately, many patients do not qualify for shortened regimens and need 48 weeks of treatment.6, 7 Data beginning to emerge since the approval of the protease inhibitors suggest that discontinuation rates from these regimens have been high.8-11 Other limitations of treatment with the available protease inhibitors are their low barrier to resistance,12 potential for drug interactions, and complex regimens with high pill burdens. Thus, a clear need exists for a shorter, simpler, better tolerated, and effective regimen with a high barrier to resistance for treatment-naive patients with chronic HCV infection.
Sofosbuvir (formerly known as GS-7977; Gilead Sciences, Foster City, CA, USA) is a selective, pangenotypic nucleotide inhibitor of NS5B-directed HCV RNA replication. In another phase 2 trial,13 43 (91%; 95% CI 80-98) of 47 treatment-naive patients with HCV genotype-1 receiving 400 mg sofosbuvir in combination with peginterferon and ribavirin for 12 weeks followed by 12 weeks of peginterferon and ribavirin had sustained virological response at post-treatment week 12 (SVR12).13 These results, along with the rapidity of the recorded on-treatment virological suppression (nearly all patients had undetectable concentrations by week 4) and the lack of viral breakthrough in this trial and other studies of sofosbuvir, including the exploratory ELECTRON phase 2 trial,14 indicate the need to assess shorter durations of treatment with sofosbuvir plus peginterferon and ribavirin in the treatment of patients with chronic HCV. The ATOMIC trial was designed to assess whether a 12-week treatment regimen of sofosbuvir plus peginterferon and ribavirin is as effective as a 24-week regimen. Additionally, we explored whether or not 12 weeks of sofosbuvir plus peginterferon and ribavirin followed by an additional 12 weeks of sofosbuvir monotherapy or sofosbuvir and ribavirin offers any benefit compared with the 12-week regimen of sofosbuvir plus peginterferon and ribavirin.
Methods
Study design and participants
We did this randomised, open-label phase 2 study at 42 centres: 41 in the USA and one in Puerto Rico. Study screening began on March 23, 2011, with the last patient enrolled on Sept 21, 2011; the last patients' final follow-up visit was on Aug 27, 2012. Eligible patients were at least 18 years of age, had not been treated previously for HCV infection, and had chronic genotype 1, 4, 5, or 6 HCV infection with serum HCV RNA concentrations of 50 000 IU/mL or greater. Exclusion criteria included histological evidence of cirrhosis (patients had to have had a liver biopsy done within 36 months of entry) or other clinically important chronic liver disease, a body-mass index of 18 kg/m2 or lower, or co-infection with hepatitis B or HIV. Patients with a history of psychiatric illness were eligible if approved by a psychiatrist or licensed mental health professional.
Before enrolment and before any procedures were done, written informed consent was obtained from all patients. The study was done in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. A safety review committee consisted of a group of Pharmasset employees (including a clinical scientist, safety scientist, medical monitor, medical advisor, and chief medical officer) who met on a monthly basis to review the ongoing safety of the study; additionally, a group of four external members (including the committee chairman) were available on an as-needed basis.
Randomisation and masking
Using an interactive web-based response system, we randomly allocated patients with HCV genotype-1 in a 1:2:3 ratio to cohorts A, B, or C. Randomisation was stratified by IL28B (CC vs non-CC allele) and HCV RNA (<800 000 IU/mL vs ≥800 000 IU/mL). Patients with genotype 4, 5, or 6 (or indeterminate genotype) were enrolled into cohort B. This study was an open-label study. For the study to have been blinded, patients in cohorts A and C would have had to receive placebo injections for 12 weeks after the conclusion of their planned dosing. We decided that the potential benefits of blinding did not warrant the risk and inconvenience to patients. Patients as well as individuals providing study treatment, assessing outcomes, or analysing data were not masked to group assignment at any point during the study.
Procedures
Individuals in cohort A received sofosbuvir 400 mg orally once daily, peginterferon 180 μg subcutaneously once a week, and ribavirin orally as a divided weight-based daily dose (ie, patients <75 kg received 1000 mg and those ≥75 kg received 1200 mg) for 12 weeks. Patients in cohort B received the same drugs at the same doses for 24 weeks. Patients in cohort C received the same regimen as individuals in cohort A followed by an additional 12 weeks of sofosbuvir monotherapy for half the patients, or sofosbuvir plus ribavirin for the other half (with patients randomly allocated to these subcohorts). Patients in cohort A who did not achieve a rapid virological response (defined as HCV RNA <15 IU/mL at week 4) continued to receive sofosbuvir plus peginterferon and ribavirin for an additional 12 weeks. After completion or early discontinuation of treatment, patients were followed up off-treatment until week 24.
We measured plasma HCV RNA concentrations using the COBAS AmpliPrep/COBAS Taqman HCV test (Roche; Indianapolis, IN, USA) with a limit of detection of 15 IU/mL. We defined virological breakthrough as the presence, during treatment, of detectable HCV RNA in serum samples after previous documentation of HCV RNA concentrations lower than 15 IU/mL; we defined virological rebound as a greater than 1 log10 increase in HCV RNA from the lowest point while on treatment. Relapse was defined as presence of detectable HCV RNA at any time during the 24-week post-treatment follow-up after documentation of HCV RNA less than 15 IU/mL in serum samples at the end of treatment. We discontinued treatment in patients who did not respond by week 12 (ie, <2 log10 decrease in HCV RNA) or who had confirmed viral breakthrough or rebound at any time during the trial.
We monitored patients for virological breakthrough during the 12-24 weeks of treatment and for relapse after treatment discontinuation. We did a confirmatory HCV RNA test in any patient with virological breakthrough and all treatment was discontinued if breakthrough was confirmed. Blood samples were obtained at each study visit for population sequencing of the NS5B-encoding region with a detection limit of 15-25% of the viral population. Samples were sequenced by dideoxy sequencing (DDL Diagnostic Laboratory; Rijswijk, Netherlands) at baseline for all patients, and at failure timepoints for those who had virological breakthrough or relapse.
For virological failures, phenotypic analysis of NS5B was done by Janssen Diagnostics BVBA (Beerse, Belgium), with a replicon-based HCV assay containing the NS5B regions of HCV derived from plasma sequences from patients and quantitatively measured differences in sofosbuvir susceptibility compared with corresponding baseline samples or respective wild-type reference replicon.
Safety was assessed by review of adverse events and concomitant drugs, blood samples for serum tests and haematological assessments, and physical examinations including vital signs and electrocardiograms. Patients with decreases in haemoglobin concentrations to lower than 100 g/L during treatment received reduced peginterferon or ribavirin dosing. The use of erythropoiesis-stimulating agents was not allowed.
Statistical analysis
The primary efficacy endpoint of the study was sustained virological response 24 weeks after discontinuation of all treatment (SVR24). The intention-to-treat analysis included all patients who were enrolled and received at least one dose of study drug. The primary analysis compared the proportion of patients in each treatment group with HCV RNA concentrations lower than 15 IU/mL (or undetectable) at week 24 after the end of treatment. We calculated point estimates and two-sided 95% CIs of between-group differences in SVR24 using stratum-adjusted Mantel-Haenszel proportions. Secondary endpoints were the proportion of patients with undetectable HCV RNA at all timepoints throughout the study (eg, rapid virological response and SVR12) with point estimates and exact 95% CIs.
We estimated that a sample size of 50 patients and 100 patients or a sample size of 75 patients and 100 patients would be sufficient to achieve 90% power to detect a 30% or 25% difference in SVR24 rates between two treatment groups with the χ2 and a 5% two-sided significance level. We used SAS (version 9.2) for all statistical analyses.
Role of the funding source
The sponsor of the study contributed to recruitment of patients, trial management, data collection, statistical analyses, and the writing and review of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
We screened 588 patients with HCV genotypes 1, 4, and 6, of whom 332 were eligible and enrolled in the study (figure 1). No patients with HCV genotype 5 were enrolled into this study. Characteristics of patients were much the same between groups at baseline, with a mean age of about 50 years and most patients being men, being white, and carrying a non-CC IL28 B genotype (table 1).
Because efficacy results for patients in cohort C who were randomly allocated into two subgroups for the second 12 weeks of treatment-those who received sofosbuvir monotherapy (cohort C1) and those who received sofosbuvir plus ribavirin (cohort C2)-were very similar, their data were pooled when assessing efficacy. However, we analysed the two subcohorts separately when assessing adverse events.
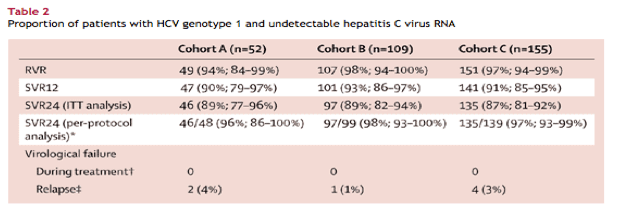
Patients in all groups had rapid and substantial reductions in HCV RNA after beginning treatment (table 2). At the end of the first week of dosing, median decreases in HCV RNA in all three cohorts were greater than 4·5 log10 IU/mL. By the second week of treatment, 79% of patients (259 of 328) receiving treatment had undetectable HCV RNA, a proportion that increased to 99% (323 of 326 patients; 97% by intention-to-treat analysis [323 of 332 patients]) at week 4 of treatment. One patient in cohort A did not have undetectable HCV RNA by week 4. Because of an administrative error, this patient did not receive an extra 12 weeks of treatment as specified by the protocol, but did achieve SVR24.
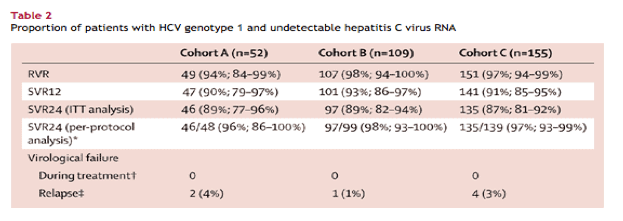
We recorded high rates of SVR12 and SVR24 in all three groups (table 2). We noted no difference in the proportions of patients achieving SVR24 between cohorts A and B (p=0·94) or between cohorts A and C (p=0·78), suggesting no additional benefit of treatment durations of longer than 12 weeks.
Of the 11 patients with genotype 4 HCV, nine (82%, 95% CI 48-98%) achieved both SVR12 and SVR24. We recorded no virological failure in these 11 patients-the other two patients were lost to follow-up at the end of treatment. All five of the patients with genotype 6 HCV achieved SVR12 and SVR24 (100%, 48-100%).
Factors shown to be associated with reduced response to treatment did not seem to greatly affect response to this regimen: rates of SVR24 for patients with high baseline HCV RNA (≥800 000 IU/mL) were 89% in cohort A (40 of 45 patients), 90% in cohort B (81 of 92 patients), and 87% in cohort C (110 of 127 patients); rates of SVR24 for patients carrying non-CC IL28B genotypes were 87% in cohort A (34 of 39 patients), 90% in cohort B (80 of 89 patients), and 88% in cohort C (96 of 116 patients); and rates of SVR24 for patients with bridging fibrosis versus those without bridging fibrosis were 100% (all seven patients) versus 87% (19 of 23 patients) in cohort A, 88% (15 of 17 patients) versus 89% (96 of 108 patients) in cohort B, and 83% (19 of 23 patients) versus 88% (116 of 132 patients) in cohort C.
No patients had viral breakthrough during treatment. Of the 11 patients who had a return of detectable HCV RNA after stopping treatment, seven relapsed after completing their assigned treatment regimen (table 2). Of these seven patients, relapse occurred by post-treatment follow-up week 4 in four patients, by follow-up week 8 in two patients, and by follow-up week 12 in one patient. All but one patient who had viral relapse carried a non-CC IL28B allele. The slightly fewer number of individuals achieving SVR12 than those receiving SVR24 in each group was not because of relapse but because of patients lost to follow-up after 12 weeks. The rate of relapse did not seem to be higher in patients who received ribavirin dose reductions during treatment (data not shown). The remaining four patients who had virological failure did not complete their assigned course of treatment. All four achieved undetectable HCV RNA during treatment, but had detectable viraemia within 8 weeks after early treatment discontinuation.
Changes in the NS5B polymerase in clinical isolates from the 11 patients who either relapsed after a full course of treatment or after early discontinuation were assessed by population sequencing. We detected neither the Ser282Thr or Met289Leu mutations at the time of virological failure. We detected no change in susceptibility to sofosbuvir compared with their corresponding baselines or in the wild-type 1b Con-1 replicon with any of the 11 patients at the time of relapse.
Most patients (97-99%) had at least one adverse event during the study. The most common adverse events were those consistent with the known safety profile for peginterferon and ribavirin: fatigue, headache, and nausea, with most of these adverse events rated by treating clinician as mild in severity (table 3). 30 patients had adverse events leading to discontinuation of any study drug. The proportion of patients with genotype-1 who discontinued any study drug because of an adverse event was greater in cohort B than in either of the other two cohorts (18% vs 5-6%; three [6%] of 52 patients in cohort A, 19 [18%] of 106 patients in cohort B, and seven [5%] of 155 patients in cohort C). The most common adverse events that led to the discontinuation of any study drug-anaemia and neutropenia-are associated with peginterferon and ribavirin treatment. Anaemia leading to dose modification or interruption seemed to be more common in cohort B (25 [20%] of 125 patients) and cohort C2 (17 [23%] of 75 patients) than it was in cohort A (five [10%] of 52 patients) and cohort C1 (eight [11%] of 75 patients), which might be a consequence of the longer treatment duration with ribavirin. Adverse events that led to treatment discontinuation in more than two patients were neutropenia, nausea, and anxiety (four patients for each), and anaemia (three patients).
13 treatment-emergent serious adverse events were reported in 12 patients: two (4%) in cohort A, six (5%) in cohort B, and four (3%) in cohort C (two each in cohorts C1 and C2). Nine serious adverse events were thought to be unrelated to study drug treatment (arrhythmia, ischaemic colitis, chest pain, acute cholecystitis, cholelithiasis, alcohol poisoning, road-traffic accident, costochondritis, and hip arthroplasty). Four serious adverse events-anaemia, autoimmune hepatitis, pyelonephritis, and pancytopenia-were reported as related to peginterferon and ribavirin (but unrelated to sofosbuvir). Two of the 13 serious adverse events (automimmune hepatitis and chest pain) led to permanent discontinuation of study drug. Subsequent testing confirmed that the case of autoimmune hepatitis was an undiagnosed pre-existing disorder. No patients died during the study period.
Across all treatment groups, we recorded a decrease in neutrophils, haemoglobin, platelets, and lymphocytes, consistent with the known effects of peginterferon and ribavirin (figure 2). The most common grade 3 or 4 laboratory abnormality was neutropenia (table 4). Recovery of neutrophil counts to baseline values occurred promptly after discontinuation of peginterferon in patients continuing on sofosbuvir or sofosbuvir plus ribavirin (figure 2). Additionally, we detected rapid increase of haemoglobin and lymphocyte counts to baseline values in patients receiving sofosbuvir monotherapy; improvement of these indices occurred at a slower rate and to a lesser extent in the group randomised to receive sofosbuvir plus ribavirin (figure 2). Although mean serum ALT concentrations decreased in all cohorts B, C1, and C2 (figure 2) during the first 12 weeks of treatment, we saw further improvements after discontinuation of peginterferon at week 12.
Discussion
Our findings suggest that sofosbuvir is well tolerated and that there is no additional benefit of extending sofosbuvir treatment beyond 12 weeks. Furthermore, patients in the groups receiving longer durations of peginterferon generally had higher rates of adverse effects without an attendant increase in efficacy.
The uniformly high rates of SVR24 with sofosbuvir plus peginterferon plus ribavirin also suggest that there would be no need to tailor either the treatment duration or regimen to individual patients on the basis of early response or baseline characteristics. Protease inhibitor regimens (approved in April, 2011) use response-guided treatment to shorten treatment duration from 48 weeks to 24-28 weeks in patients who fulfil predefined criteria for early response.2, 3 Our results indicate that response-guided treatment might not be needed for treatment with sofosbuvir. Moreover, on the basis of our findings, other factors previously shown to be predictive of response to treatment-IL28B CC versus non-CC genotype, high versus low baseline viral load, and genotype 1a versus genotype 1b-are of doubtful use with this sofosbuvir regimen. Although all but one patient who relapsed carried non-CC IL28B alleles, the predictive value of this index for response seems insufficient to base treatment decisions on. However, the small numbers of patients in some of these subgroups do not allow definitive conclusions.
The advent of direct-acting antivirals has been accompanied by concerns about the development of drug resistance.7 In phase 3 trials of the protease inhibitors telaprevir and boceprevir, resistance-associated mutations were detected in up to 75% of patients who did not achieve SVR.12 More than 90% of patients who had virological failure in these trials were shown to harbour drug-resistant variants.12 Our findings seem to lend support to the claim that sofosbuvir has a high barrier to resistance. We detected no virological breakthrough or treatment-emergent resistance in this trial. Relapse after treatment was rare, and no patients who had relapse showed the presence of the signature Ser282Thr mutation in population sequencing.
Sofosbuvir-based treatment seemed to be safe and well tolerated. Most of the adverse events and laboratory abnormalities seen during this study were characteristic of peginterferon or ribavirin. Patients in cohort C who received sofosbuvir monotherapy after 12 weeks of triple therapy showed prompt improvement in haemoglobin and neutrophil values towards baseline values, suggesting little or no haematological toxicity that could be ascribed solely to sofosbuvir. Patients in the third cohort who received sofosbuvir and ribavirin combination treatment after 12 weeks of triple therapy also showed improvement in haematological indices, although recovery was slower, consistent with the effect of ribavirin.
In terms of its rate of response, resistance profile, and safety characteristics, sofosbuvir plus peginterferon plus ribavirin for 12 weeks seems to compare favourably with those seen with present standard-of-care treatments for treatment-naive patients with HCV genotype-1 (panel).
------------------------------
Panel
Research in context
Systematic review
We consulted a review of treatment of hepatitis C in adults,14 which systematically assessed a large body of evidence concerning outcomes of clinical trials of approved drug regimens for the treatment of hepatitis C virus (HCV). We searched PubMed in December, 2012, using the search term "HCV treatment", searching for studies written in English. We also consulted treatment guidelines for hepatitis C.1, 15
Interpretation
For previously untreated patients with genotype-1 hepatitis C infection, standard-of-care treatment is one of the recently approved (April, 2011) protease inhibitors-telaprevir or boceprevir-plus peginterferon and ribavirin. Our findings lend support to the phase-3 assessment of a 12 week sofosbuvir regimen in a broader population of patients with chronic HCV genotype-1 infection, including those with cirrhosis. Furthermore, the exploration of the combination of sofosbuvir with other direct-acting antiviral agents is warranted.
----------------------------
Sofosbuvir-based combination treatment also seemed to be effective in patients with HCV genotypes 4 and 6; however, the small numbers of patients in the study with genotype 4 and 6 preclude any definitive conclusions. Nor do our data allow us to address whether 12 weeks of treatment is sufficient for patients with these genotypes, because all patients with genotypes 4 or 6 received 24 weeks of triple therapy. However, we saw no breakthrough or relapse in any patient with HCV genotype 4 or 6. The only two patients (both genotype 4) who did not achieve SVR24 were lost to follow-up.
This study was limited by its exclusion of patients that are historically more difficult to treat-namely, those with cirrhosis and advanced liver disease. Ongoing phase 3 trials (NCT01497366, NCT01641640, NCT01542788, and NCT01604850) of sofosbuvir include patients with cirrhosis. We have planned future studies to examine the effectiveness of this agent in patients who have failed to respond to telaprevir-based or boceprevir-based combination treatment.
Our findings suggest that simple, short sofosbuvir-based regimens are effective for patients with HCV genotypes 1, 4, and 6. Further study of this agent in phase 3 studies are warranted and ongoing.
|
|
| |
| |
|
|
|