| |
Interferon-a as a "Curative Strategy" or Harmful?
|
| |
| |
Download the PDF here
Download the PDF here
Download the PDF here
There were 2 oral presentations on IFN as a potential therapy in the same oral session, link below, in the first talk the authors hypothesize how IFN could be used to reduce HIV reservoir & in a curative strategy, its associated with elite controllers, they mention they are pursuing HIV persistence & eradication research with HIV host restriction genes & IFN:
http://webcasts.retroconference.org/console/player/19424?mediaType=podiumVideo
Extended Interferon-Alpha Therapy Accelerates Telomere Length Loss in Human Peripheral Blood T Lymphocytes.....PLoS One.....August 4, 2011......HALT-C Trial Group.....diminished naive T cell proliferative TL reserve incurred under sustained IFN therapy may persist well beyond the end of therapy
http://www.natap.org/2011/HCV/080611_03.htm
Interferon & Aging/Inflammation/Activation:
http://www.natap.org/2012/CROI/croi_86.htm
Interferon-alpha & Inflammation: interferon stimulates activation/inflammation, therapies to quell activation....."There's a large body of evidence that excessive production of IFN-α may underlie the inflammation in [autoimmune disorder] Lupus; Lupus patients also tend to develop early-onset heart disease....."If we can understand more about the mechanisms we can potentially do something to suppress this ongoing immune activation and inflammation, and hopefully prevent the early onset of these aged-related diseases."....."We found that the group with poorer immune recovery showed evidence of increased IFN-α activity in their immune cells, we believe that cell recovery may be adversely affected by the effects of IFN-α,"....."If we can understand more about the mechanisms we can potentially do something to suppress this ongoing immune activation and inflammation, and hopefully prevent the early onset of these aged-related diseases."
Effects of Interferon-α Treatment on Anti-HIV-1 Intrinsic Immunity in vivo: "Taken within the context of recent data demonstrating that IFN-a decreases the size of the HIV-1 reservoir, intrinsic immune factors may contribute to therapeutic and curative strategies for HIV-1 infection."
http://www.retroconference.org/2013b/Abstracts/47680.htm
M Abdel-Mohsen1, T Liegler1, J Guatelli2, M Salama3, H Ghanem3, A Rauch4, B Ledergerber5, H Gunthard5, J Wong1,6, Satish Pillai*1,6, and Swiss HIV Cohort
Study
1Univ of California, San Francisco, US; 2Univ of California, San Diego, US; 3Ain Shams Univ, Cairo, Egypt; 4Univ Hosp Berne, Switzerland; 5Univ Hosp Zurich, Switzerland; and 6VAMC, San Francisco, CA, US
Blockade of Type I Interferon during Acute SIV Infection Results in Accelerated Progression to AIDS and Death
http://www.retroconference.org/2013b/Abstracts/46260.htm
Netanya Sandler*1, R Zhu1, J Estes2, E Boritz1, J Lifson2, D Levin3, J Langer4, G Schreiber3, S Rao1, and D Douek1
1NIH, Bethesda, MD, US; 2NIH, Frederick, MD, US; 3Weizmann Inst of Sci, Rehovot, Israel; and 4Univ of Med and Dentistry of New Jersey and Robert Wood Johnson Med Sch, Piscataway, US
Conclusions: Blockade of type I IFN signaling during SIV exposure and acute SIV infection resulted in accelerated progression to AIDS and death, suggesting that type I IFN are critical for early control of damage to the immune system. Studies are underway to assess the effects of type I IFN during SIV exposure and acute SIV infection
UCLA press release:
Blocking a key protein boosts immune system's ability to clear chronic infection
'Entirely illogical' finding from UCLA study suggests potential therapy for HIV
By Elaine Schmidt April 11, 2013
http://newsroom.ucla.edu/portal/ucla/blocking-a-key-protein-boosts-245145.aspx
---------------
Science 12 April 2013:
Perspective
An Interferon Paradox
Pamela M. Odorizzi and E. John Wherry
Institute for Immunology and Department of Microbiology,
University of Pennsylvania Perelman School of Medicine
Type 1 interferons (IFN-α/ß) are a major first line of host defense against viral infection. Because of this potent antiviral activity, IFN-based therapies have been developed for chronic infections with hepatitis B and C viruses, as well as for HIV. However, a poorly understood phenomenon has been the persistence of virus despite induction of antiviral immune responses by type 1 IFNs. On page 207 and 202 in this issue, Teijaro et al. (1) and Wilson et al. (2) address this long-standing question and find that IFN-α/ß can also suppress the immune system in ways that promote viral persistence. This paradoxical finding should spur a reassessment of the fundamental roles of IFN-α/ß during chronic infections.
In the early stage of viral infection, recognition of pathogen-associated molecular patterns (PAMPs) through molecular sensors in the body, such as Toll-like receptors, leads to rapid production of type 1 IFNs by various cell types (3, 4). IFN-α/ß acts in both an autocrine and paracrine manner to induce the expression of IFN-stimulated genes that limit viral replication and spread (4, 5). Importantly, loss of IFN-α/ß signaling in animal models usually leads to uncontrolled viral replication (6). Chronic viral infections can result in sustained IFN-α/ß signaling, presumably due to ongoing recognition of viral PAMPs (7). It has been unclear, however, why this ongoing IFN-α/ß signaling during chronic infections does not lead to viral control.
Teijaro et al. and Wilson et al. used a mouse model of chronic infection with either of two strains of lymphocytic choriomeningitis virus (LCMV)-the Armstrong strain, which is associated with T cell-mediated viral control, and the clone 13 strain, which induces broad immune dysfunction, including T cell exhaustion (gradual decrease in T cell function). Both strains trigger robust, but transient, IFN-α/ß production; however, the expression of many IFN-stimulated genes persists during chronic infection. In these studies, the removal of IFN-α/ß signaling in animals through genetic deletion of a subunit for the type 1 IFN receptor, or antibody-mediated blockade of the IFN receptor before infection, increased viral replication and acute LCMV infection was no longer controlled. Surprisingly, inhibiting IFN-α/ß signaling also reduced the expression of immunosuppressive molecules, such as the regulatory cytokine interleukin-10 (IL-10) and the inhibitory receptor ligand programmed death ligand 1 (PD-L1). The IL-10 and PD-1 pathways promote viral persistence and T cell exhaustion during many chronic viral infections (8). Despite an initial increase in virus, blocking IFN-α/ß during chronic LCMV infection led to a substantial reduction in viral titers by 2 months after infection. Therapeutic blockade of IFN-α/ß signaling with an antibody against the IFN receptor after the establishment of chronic infection also enhanced viral control. Both studies observed improved virus-specific CD4+ T cell responses and preserved lymphoid tissue organization in the absence of IFN-α/ß signaling.
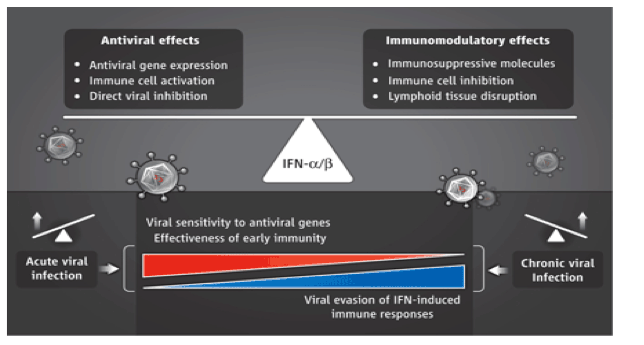
Balancing dual roles.
Type 1 interferons (IFN-α/ß) may control viral replication and spread through two mechanisms. Antiviral responses include the expression of antiviral genes and the activation of specific immune cells. Immunomodulatory responses include the expression of immunosuppressive molecules, immune cell inhibition, and cell death. The balance of these responses may shift, with enhanced antiviral actions during acute infections and greater immunomodulatory effects during chronic infections.
The studies by Teijaro et al. and Wilson et al. highlight an unappreciated dual nature of IFN-α/ß signaling during chronic viral infections (see the figure).
Type 1 IFNs limit early viral replication through multiple direct molecular mechanisms, including inhibition of viral transcription and translation, as well as degradation of viral nucleic acids (5). The production of IFN-α/ß early upon infection also serves as an activation signal for immune cells by promoting lysis of infected cells by natural killer cells, enhancing antigen presentation-T cell priming by dendritic cells, and sustaining proliferation and activation of T cells (3-5). However, IFN-α/ß also has regulatory effects that suppress immune responses. For example, IFN-α/ß can trigger programmed cell death of activated T cells and increase production of immunosuppressive molecules, including IL-10, PD-L1, and indoleamine (2,3)-dioxygenase (4, 9). Thus, although early antiviral effects of IFN-α/ß are critical, the potential immunoregulatory roles of IFNs later in chronic infection could explain paradoxical clinical observations using IFN-based treatments. For example, strong basal IFN-α/ß signatures (increased expression of IFN-stimulated genes) correlate with poor responses to IFN-α therapy during chronic hepatitis C virus infection (10, 11). Such signatures are also associated with disease progression during chronic HIV and pathogenic SIV infections, as well as during active versus latent Mycobacterium tuberculosis infection (12-14).
Why might IFNs elicit direct antiviral effects while concurrently boosting immunoregulatory responses that prevent robust adaptive immune responses to infections? One possibility is that the immunoregulatory functions of IFN-α/ß may have evolved to limit immune-mediated pathology during infections where viral persistence is inevitable. In these settings, the IFN-α/ß pathway may sense the level of ongoing viral replication and bolster immune suppression to avoid damaging immunopathology, such as a "cytokine storm" (uncontrolled cytokine production and immune cell activation), meningitis, or immune-mediated tissue destruction. An essential next step will be determining how this balance is influenced by pathogen virulence and the strength of the immune response. In addition, the ability of IFN-α/ß to efficiently control early viral replication, and the capacity of different viruses to evade this response, may dictate the importance of these immunoregulatory effects.
How might these findings improve IFN-based treatment strategies? There are several issues to be addressed. Identifying the molecular basis for the antiviral versus immunomodulatory effects of IFN-α/ß will be necessary to selectively manipulate these opposing activities. It will also be critical to determine how the balance between antiviral and immunoregulatory effects varies from virus to virus or during a single viral infection over time. The studies of Teijaro et al. and Wilson et al. suggest that patients currently on IFN therapy could be monitored for the induction of antiviral versus immunoregulatory effects, allowing physicians to modify treatment strategies accordingly. It also may be possible to further improve the antiviral therapeutic potential of IFNs and perhaps exploit the immunoregulatory properties of this pathway for nonviral diseases.
----------------------------
Science 12 April 2013
Elizabeth B. Wilson et al.
http://www.sciencemag.org/content/340/6129/202.full
Blockade of Chronic Type I Interferon Signaling to Control Persistent LCMV Infection
Author's Summary
INTERFER(ON)ing Persistence
During persistent viral infections, a dysregulated immune response fails to control the infection. Wilson et al. (p. 202) and Teijaro et al. (p. 207; see the Perspective by Odorizzi and Wherry) show this occurs because type I interferons (IFN I), critical for early responses to viral infection, contribute to the altered immunity seen during persistent infection. Antibody blockade of IFN I signaling during chronic lymphocytic choriomeningitis virus (LCMV) in mice resulted in reduced viral titers at later stages of infection, reduced expression of inhibitory immune molecules and prevented the disruptions to secondary lymphoid organs typically observed during persistent infection with LCMV. Whether type I IFNs are also detrimental to persistent viral infection humans, such as HIV and hepatitis C virus, remains to be determined.
ABSTRACT
Type I interferons (IFN-I) are critical for antiviral immunity; however, chronic IFN-I signaling is associated with hyperimmune activation and disease progression in persistent infections. We demonstrated in mice that blockade of IFN-I signaling diminished chronic immune activation and immune suppression, restored lymphoid tissue architecture, and increased immune parameters associated with control of virus replication, ultimately facilitating clearance of the persistent infection. The accelerated control of persistent infection induced by blocking IFN-I signaling required CD4 T cells and was associated with enhanced IFN-γ production. Thus, we demonstrated that interfering with chronic IFN-I signaling during persistent infection redirects the immune environment to enable control of infection.
Despite initially robust antiviral immune activity, some viruses, including HIV and hepatitis C virus (HCV) in humans and lymphocytic choriomeningitis virus (LCMV) in mice, outpace the immune response and establish persistent infections (1, 2). Besides virus-mediated evasion tactics, the host initiates an immunosuppressive program that actively suppresses antiviral T cell responses and facilitates persistent infection (3-8). The expression of suppressive factors is tightly linked to viral burden (3, 4, 8), suggesting the presence of an immunologic sensory system that continually measures the magnitude and duration of viral replication and then dynamically modulates the balance between antiviral immunity and immune exhaustion.
In order to identify the mechanisms orchestrating the immunosuppressive program during virus infection, we performed RNA microarray-based splenic network analysis. We compared mice infected with one of two LCMV strains: the Armstrong (Arm) strain, which induces a robust T cell response that resolves infection within 8 to 10 days, or the clone 13 (Cl13) strain, which generates a persistent infection because of the sustained expression of an immunosuppressive program, including production of interleukin (IL)-10 and expression of the inhibitory molecule PD-L1 (programmed cell death 1 ligand 1) (4, 5, 9-13). PD-L1 and IL-10 are similarly expressed at the onset of both acute and persistent infection; however, expression of these molecules wanes with resolution of acute infection, whereas they are maintained or elevated in persistent infection (3, 4, 8).
Similarly, antigen-presenting cell (APC) populations expressing multiple suppressive factors with the ability to inhibit T cell responses are present early in acute infection but are elevated in the context of persistent infection (8). We focused our microarray analysis to identify factors exhibiting a similar kinetic that might be used to sense virus replication dynamics and control immunosuppressive programs. Tissue-wide cytokine expression patterns were similar in acute and persistent infections (fig. S1A). However, analogous to virus clearance kinetics, type I interferon (IFN-I) receptor (IFNR)-stimulated genes, signal transducer and activator of transcription (STAT) genes, and IFN-I regulatory factors were initially similarly expressed in LCMV-Arm and LCMV-Cl13 infections but then rapidly dissipated as acute LCMV-Arm infection resolved, whereas they remained elevated in LCMV-Cl13 infection (Fig. 1A and table S1). In total spleen, the expression of IFN-α and IFN-ß was not elevated above uninfected mice (fig. S1B); however, at day 9 postinfection IFN-α and IFN-ß transcripts were still present in dendritic cells (DCs) from LCMV-Cl13-infected mice (Fig. 1B). Analysis of IL-10-green fluorescent protein (GFP) (Vert-X) reporter mice (8, 14) revealed that OAS and Mx1 (genes directly stimulated by IFNR signaling) expression levels were specifically enriched in the immunoregulatory APCs that coexpress the highest levels of PD-L1 and IL-10 and can suppress antiviral T cell responses (8) (Fig. 1C), suggesting a link between prolonged IFN-I signaling and immunosuppression. Expression of IRF3, a gene involved in the IFN-I response but whose expression is not directly regulated by IFNR signaling (15), was not differentially increased in immunoregulatory APCs (Fig. 1C).
We next determined the impact of IFN-I signaling on the immunosuppressive program in vivo. Although levels of virus replication usually correlate with expression of suppressive factors (1), Ifnar1-/- mice exhibited decreased expression of PD-L1 and IL-10 compared with wild-type mice at day 9 after LCMV-Cl13 infection (Fig. 2A and fig. S2A) despite elevated levels of viral replication and viral antigen (fig. S2, A and B). Furthermore, treatment of splenocytes with IFNß stimulated increased PD-L1 and IL-10 expression (fig. S2C). Taken together, these data suggest that IFN-I signaling drives the immunosuppressive program in vivo. Ifnar1-/-mice failed to clear LCMV-Arm by day 9 after infection (fig. S2D), consistent with the antiviral and immune stimulatory effect of IFN-I during viral infection (16).
To resolve the role of IFN-I in induction of the immunosuppressive program separate from potential abnormalities of life-long genetic deficiency in Ifnar1-/- mice (17), we treated wild-type mice with an IFNR1-blocking antibody beginning 1 day before LCMV-Cl13 infection. IFNR1 blockade diminished Mx1, OAS, and IRF7 expression in multiple tissues and cell types (fig. S3A), indicating the ability to inhibit IFN-I signaling in vivo. Analogous to persistently infected Ifnar1-/- mice, IFNR1 antibody blockade led to decreased PD-L1 and IL-10 expression and elevated virus titers compared with isotype antibody-treated LCMV-Cl13-infected mice on day 9 after infection (Fig. 2B). IL-10 levels rebounded when IFNR1 blocking antibody treatment was withdrawn (day 15; Fig. 2B), indicating sensitive surveillance and rapid modulation of the immunosuppressive state through IFN-I signaling. Heightened IFN-I signaling can inhibit inflammasome activity in some situations (18). However, despite higher levels of virus replication and LCMV antigen in splenic APCs from persistently infected mice treated with IFNR1 blocking antibody (Fig. 2B and fig. S2B), reduced amounts of IL-1, IL-18, and inflammasome activation were observed (fig. S3B), indicating that blockade of IFN-I signaling decreases chronic inflammation during persistent infection. The reduced levels of inhibitory factors and chronic activation after IFNR1 blockade were not indicative of global down-regulation of proinflammatory cytokines, and in fact expression of IFN-γ, a factor critical for control and therapeutic resolution of persistent LCMV infection (16, 19, 20), was elevated after IFNR1 blockade (fig. S3B). Thus, IFNR blockade during persistent infection diminishes immunosuppressive signals and chronic inflammation during persistent infection.
IFNR1 antibody blockade before infection also decreased the level of IL-10- and PD-L1-expressing immunoregulatory DCs (Fig. 2C), leading to an enhanced ratio of stimulatory to immunoregulatory DCs. Moreover, IFNR1 antibody blockade prevented the splenic disorganization associated with impaired immune cell interactions and the inability to control persistent infection (21-23) (Fig. 2D and fig. S3C). Thus, immune cells are likely better positioned to interact with one another, and, because of the decrease in immunoregulatory DC frequency, those interactions are more likely to be stimulatory.
We next determined how blockade of IFN-I signaling before infection contributed to control of persistent infection. Although virus titers were initially increased in mice treated with IFNR1 blocking antibody, by 30 days postinfection viremia was reduced compared with isotype treatment, and many of the mice had already controlled infection (Fig. 3A). Furthermore, virus titers were decreased in multiple compartments, including the kidney (a life-long reservoir of LCMV-Cl13) (Fig. 3B). Mirroring findings in Ifnar1-/- mice (16), IFNR1 blockade in wild-type mice led to persistent infection with LCMV-Arm (fig. S2D), demonstrating the antiviral activity of IFN-I and its requirement to control acute viral infection.
We next sought to understand the immune mechanisms through which IFNR1 blockade enables control of persistent viral infection. IFNR1 blockade before infection induced a numerical increase in many immune subsets 9 days after infection including the total number of functional virus-specific CD4 T cells (Fig. 3C and fig. S4A). However, despite an overall increase in B cells and CD4 T cells, LCMV-specific antibody titers were not elevated in IFNR1 blocking antibody-treated mice at day 9 or 30 after infection (fig. S4B). Unlike virus-specific CD4 T cells, virus-specific CD8 T cell numbers and cytokine production were similar or slightly reduced when IFN-I signaling was blocked (Fig. 3C and fig. S4C). On the basis of the increase in natural killer (NK) cells, virus-specific CD4 T cells, and systemic IFN-γ levels (Fig. 3C and figs. S3B and S4A), we sought to examine the role of each of these factors in accelerating virus clearance after IFNR1 blockade. CD4 depletion before infection abrogated the accelerated virus control engendered by IFNR1 blockade, whereas NK cell depletion did not affect IFNR1 blockade-mediated clearance (Fig. 3D). To assess the role of increased IFN-γ in IFNR1 blockade-induced virus clearance, we treated mice with IFNR1 blocking antibody with or without IFN-γ blocking antibodies at the time of LCMV-Cl13 infection. The accelerated clearance of persistent infection after IFNR1 blockade was abrogated in mice cotreated with IFN-γ blocking antibody (anti-IFN-γ) (Fig. 3E). Although occurring later than in IFN-γ knockout mice (24), mice treated with anti-IFN-γ alone died ~35 days after infection, whereas mice receiving IFNR1 blocking antibody plus anti-IFN-γ survived and cleared infection similar to untreated mice. Together, these results indicate that IFNR1 antibody stimulates accelerated clearance of persistent virus infection through CD4 T cell and IFN-γ-dependent mechanisms.
We next determined whether therapeutic blockade of IFN-I signaling affected an established LCMV-Cl13 infection. Blockade of IFNR1 beginning 25 days after infection accelerated control of persistent infection in multiple compartments compared with isotype treatment (Fig. 4, A and B). The enhanced control of infection occurred despite the initial increase in virus titers immediately after IFNR1 blocking antibody therapy (Fig. 4A). Blockade of IFNR1 beginning 25 days after infection reduced the IFN-I gene expression signature and decreased IL-10 and PD-L1 levels (Fig. 4, C and D), demonstrating that IFN-I continues to be a key component of an immunologic surveillance system and stimulator of the immunosuppressive program throughout persistent infection. Thus, therapeutically ablating chronic IFN-I immune activation in vivo enhances control of persistent LCMV infection.
Our results demonstrate that chronic IFN-I signaling during persistent infection drives the immunosuppressive program and that interfering with IFN-I signaling restores multiple parameters of productive immunity, allowing for viral clearance. IFN-I treatment in combination with the antiviral drug ribavirin is often effective at eradicating HCV infection. However, some patients fail to have a sustained virologic response. A characteristic of patients that fail IFN-I/ribavirin therapy is a heightened IFN-I signature before treatment that fails to substantially increase with therapy (25, 26). Thus, the initially high IFN-I signature may lead to enhanced immune dysfunction, and consequently adding more IFN-I is ineffective. These results highlight the duality of IFN-I during viral infection: Acute IFN-I signals possess antiviral and immune stimulatory potential required for clearance of infection, but when virus cannot be controlled, acutely sustained IFN-I signaling induces immunosuppression that facilitates persistent virus infection. Considering that HIV and HCV infections are also associated with immune activation driven by chronic IFN-I signaling (23, 27-29), a similar blockade of IFN-I may improve control of these infections. In total, our data support IFN-I as a central rheostat and regulator of the immunosuppressive program and the possibility that it may be feasible to redirect entire immunologic programs by modulating activity of a single pathway: IFN-I.
----------------------------
Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling
Science 12 April 2013
http://www.sciencemag.org/content/340/6129/207.full
John R. Teijaro et al.
Editor's Summary
INTERFER(ON)ing Persistence
During persistent viral infections, a dysregulated immune response fails to control the infection. Wilson et al. (p. 202) and Teijaro et al. (p. 207; see the Perspective by Odorizzi and Wherry) show this occurs because type I interferons (IFN I), critical for early responses to viral infection, contribute to the altered immunity seen during persistent infection. Antibody blockade of IFN I signaling during chronic lymphocytic choriomeningitis virus (LCMV) in mice resulted in reduced viral titers at later stages of infection, reduced expression of inhibitory immune molecules and prevented the disruptions to secondary lymphoid organs typically observed during persistent infection with LCMV. Whether type I IFNs are also detrimental to persistent viral infection humans, such as HIV and hepatitis C virus, remains to be determined.
ABSTRACT
During persistent viral infections, chronic immune activation, negative immune regulator expression, an elevated interferon signature, and lymphoid tissue destruction correlate with disease progression. We demonstrated that blockade of type I interferon (IFN-I) signaling using an IFN-I receptor neutralizing antibody reduced immune system activation, decreased expression of negative immune regulatory molecules, and restored lymphoid architecture in mice persistently infected with lymphocytic choriomeningitis virus. IFN-I blockade before and after establishment of persistent virus infection resulted in enhanced virus clearance and was CD4 T cell-dependent. Hence, we demonstrate a direct causal link between IFN-I signaling, immune activation, negative immune regulator expression, lymphoid tissue disorganization, and virus persistence. Our results suggest that therapies targeting IFN-I may help control persistent virus infections.
Persistent viral infections such as HIV, hepatitis B virus, and hepatitis C virus (HCV) represent important global health problems. Persistent viruses take advantage of negative immune regulatory molecules to suppress antiviral CD4 and CD8 T cell responses (1, 2), resulting in T cell exhaustion (3, 4) and facilitating virus persistence. Hyperimmune activation is also observed after persistent virus infection and is characterized by prolonged activation of T cells, B cells, and natural killer (NK) cells; elevated pro-inflammatory mediators; and a sustained interferon signature (5-7). Type I interferon (IFN-I) signaling is upstream of hundreds of inflammatory genes, suggesting that IFN-I may be responsible for generating the hyperactivated immune environment during virus persistence. We investigated the role of IFN-I in regulating immune activation, immune suppression, and virus control after persistent virus infection in mice.
To elucidate the role of IFN-I in virus persistence, we used lymphocytic choriomeningitis virus (LCMV). In adult mice, the Armstrong (Arm) strain causes an acute infection that is cleared 8 days postinfection (dpi) because of robust antiviral CD8 T cell responses. In contrast to the Arm strain, the clone 13 (Cl13) strain causes a systemic viral infection lasting over 90 days (8-13).
Cl13-infected mice had significantly elevated IFN-I in the serum compared with Arm-infected counterparts at 18 and 24 hours postinfection (hpi) (Fig. 1, A and B). By using IFN-ß-yellow fluorescent protein (YFP) reporter mice (14), we detected YFP expression in plasmacytoid dendritic cells (pDCs) at 18 hours post-Cl13 infection, with minimal YFP expression in pDCs during Arm infection (fig. S1A). IFN-ß-YFP expression was not observed in other splenocytes (fig. S1B), suggesting that Cl13 infection induces IFN-ß production in pDCs. pDCs are reported to be an early target of Cl13 infection (13, 15). To address whether Cl13 preferentially infected pDCs, we used nonreplicating Arm or Cl13 viruses, in which their glycoproteins (GPs) were replaced with a green fluorescent protein (GFP) marker (denoted ∼GP-Cl13 or ∼GP-Arm). As expected, pDCs exhibited a 2- to 2.5-fold increase in GFP expression upon infection with ∼GP-Cl13 compared with ∼GP-Arm (Fig. 1C). Consistent with IFN-I signaling being upstream of inflammatory gene expression, we observed elevated expression of multiple pro-inflammatory cytokines and chemokines 18 hours post-Cl13 infection versus Arm infection (fig. S1C). To determine whether elevated pro-inflammatory cytokines and chemokines in Cl13 infection were due to IFN-I signaling, we treated mice with an antibody against interferon alpha-beta receptor 1 (anti-IFNAR1) before infection and measured cytokine and chemokine levels in the serum 18, 24, and 48 hpi (16). Blockade of IFN-I signaling significantly blunted production of multiple pro-inflammatory cytokines and chemokines after Cl13 infection at 18, 24, and 48 hpi (fig. S1, C to E).
We asked whether IFN-I signaling contributes to the Cl13-induced immunosuppressive state. The IFN-I signaling blockade resulted in significant suppression of interleukin-10 (IL-10) production 1 and 5 dpi (Fig. 2A). We also detected significant suppression of programmed cell death 1 ligand 1 (PD-L1) on both CD8α+ and CD8α- DCs 1 dpi (Fig. 2B), which was retained 5 and 9 dpi in CD8α- but not CD8α+ DCs (Fig. 2, C and D). Together, these results demonstrate that IFN-I signaling inhibits negative regulatory molecule expression. Because DCs are primary targets of Cl13 infection and DC infection is crucial for virus persistence (8, 17, 18), we asked whether blockade of IFN-I signaling altered the DC compartment. IFN-I blockade increased virus nucleoprotein (NP) expression in DCs and macrophages 5 dpi (fig. S2C). Blockade of IFN-I signaling significantly increased both the frequency and number of CD8α- and CD8α+ DCs and macrophages (fig. S2A). Moreover, we observed a significant increase in DCs with an immune-stimulatory phenotype after blockade of IFN-I signaling (fig. S2B).
The regulation of IL-10 and PD-L1 expression by IFN-I led us to investigate how IFN-I affects the immune environment during persistent virus infection. IFN-I blockade before Cl13 infection resulted in increased splenocyte numbers in anti-IFNAR1 compared with control treated mice 9 dpi (fig. S3A). This correlated with significant increases in B cells, CD4 and CD8 T cells, NK cells, DCs, and macrophages (fig. S3, B and C). Although IFN-I blockade resulted in early inhibition of multiple pro-inflammatory cytokines and chemokines and negative immune regulatory molecules after Cl13 infection (Fig. 2 and fig. S1, C to E), we detected increases in IFN-γ production 24 hpi (fig. S2D) and similar levels of pro-inflammatory cytokines and chemokines 5 dpi (fig. S3D).
Lymphoid architecture is integral to induction and maintenance of immune responses (19-23). Cl13 infection resulted in severe lymphoid disorganization (23) with indistinguishable marginal zones and follicular structures and scattered B and T cell zones 9 dpi (Fig. 2E), which was more apparent at 14 dpi (Fig. 2F). IFN-I blockade preserved splenic architecture, so that white pulp, follicle margins, and T and B cell zones appeared similar to naïve spleens (Fig. 2E, middle and bottom). Fibroblastic reticular cell staining (ER-TR7; Fig. 2E, middle row) highlighted preservation of splenic organization and architecture after IFN-I blockade. These data demonstrate that IFN-I signaling contributes to splenic architecture disorganization during Cl13 infection.
We next asked whether blockade of IFN-I signaling altered control of Cl13. IFN-I blockade resulted in increased percentages of lymphocytes expressing LCMV viral antigen 24 hpi (fig. S2, C and D) and significantly higher Cl13 titers in the serum 10 dpi (Fig. 3A), suggesting that anti-IFNAR1 antibody treatment blocked early antiviral effects of IFN-I. By 30 dpi, we observed significant reductions in Cl13 titers after IFN-I blockade (>1.5 logs) compared with isotype control treated mice (Fig. 3A). By 40 dpi, IFN-I blockade resulted in significant reductions of viral titers in both serum and tissues (Fig. 3B). By 50 dpi, virus was undetectable in the serum after IFN-I blockade, whereas control mice retained >3 logs of virus (fig. S4A), demonstrating that IFN-I blockade hastens clearance of Cl13 infection.
IFN-I transcripts are detectable in DCs several weeks after Cl13 infection (24). We postulated that blocking IFN-I signaling during an established Cl13 infection would result in faster viral clearance. After an initial spike in viral titers 20 dpi, we observed >1-log reduction in serum viral titers in anti-IFNAR1 compared with isotype-treated mice by 40 dpi (fig. S4B). By 50 dpi, 75% of the anti-IFNAR1 treated mice had undetectable levels of virus, whereas 75% of control animals maintained >3 logs of virus (Fig. 3D). Analysis of virus in liver and lung 50 dpi revealed reductions in viral titers in both tissues after IFN-I blockade (Fig. 3D). These results demonstrate the therapeutic potential of IFN-I signaling blockade.
We asked whether enhanced virus clearance after IFN-I blockade could be duplicated after Arm infection. IFN-I blockade during Arm infection resulted in significantly elevated viral titers in the serum compared to control mice (fig. S5A). Anti-IFNAR1 treated animals maintained >3 logs of virus in serum 20 dpi (fig. S5B). Moreover, after IFN-I blockade, viral titers were detectable in lungs, kidneys, and brains 30 dpi, a time when virus was undetectable in tissues of control mice (fig. S5C). The inability to clear Arm correlated with reduced expansion, functional potential, and cytolytic capacity of LCMV-specific CD8 T cells (fig. S5, D to G) with minimal effects on LCMV-specific CD4 T cells (fig. S5H). Clearance of Arm infection relies solely on antiviral CD8 T cells; thus, inhibition of IFN-I antiviral effects coupled to abrogation of CD8 T-cell responses likely contributed to defective control of Arm infection.
To measure localization of naïve T cells to T cell zones in the spleen, we adoptively transferred carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled naïve T cells into Cl13-infected mice treated with isotype antibodies or anti-IFNAR1. Naïve T cells migrated to T cell zones in anti-IFNAR treated mice similar to naïve controls 5 dpi. Although T cell zones were intact in isotype-treated mice, naïve T cells did not remain in these areas (Fig. 4, A and B) despite similar numbers of naïve CFSE-labeled T cells in the spleen. At 14 dpi, differences in naïve T cell localization between anti-IFNAR1 and isotype control treated mice were maintained (Fig. 4B). Analysis of virus-specific T cell function revealed that the numbers of GP33-specific IFN-γ+ or IFN-γ+ tumor necrosis factor-α (TNF-α+) IL-2+ multifunctional cytokine-producing cells (Fig. 4C) along with cytolytic potential (fig. S6A) after anti-IFNAR1 treatment were comparable to those in isotype control treated mice, whereas there was a significant decrease in IFN-γ+ TNF-α+ GP33-specific CD8 T cells (Fig. 4C). In contrast, GP61-specific IFN-γ+ and multifunctional CD4 T cells 9 dpi were elevated in anti-IFNAR1 compared with control treated mice (Fig. 4D). Despite elevated numbers and enhanced functional potential of virus-specific CD4 T cells, we observed similar levels of LCMV-specific immunoglobulin G (IgG) in the serum (fig. S6B), demonstrating that IFN-I blockade enhances virus-specific CD4 T cell responses while maintaining antiviral CD8 T cell and antibody levels.
Because blockade of IFN-I signaling resulted in significantly elevated virus-specific CD4 T cell responses, we asked whether CD4 T cells were required for virus control after IFN-I blockade. Antibody depletion of CD4 T cells had little effect on anti-IFNAR1-mediated reduction of viral titers on day 21 postinfection (Fig. 4E); however, by 40 and 50 dpi, CD4 depletion completely abrogated the anti-IFNAR1-mediated reduction in viral titers compared with CD4-sufficient, IFNAR1-treated mice (Fig. 4, E and F). Anti-IFNAR1 treatment after CD4 depletion had no effect on controlling Cl13 replication in lungs, kidneys, and brains 75 dpi (Fig. 4G). These data demonstrate CD4 T cells are required for enhanced control of persistent virus infection after IFN-I blockade.
We identify IFN-I signaling as essential for immune activation, up-regulation of negative immune regulators, lymphoid disorganization, and virus persistence.
IFN-I has pleiotropic effects on multiple cellular processes. Aside from antiviral effects (25), IFN-I signaling influences cell differentiation, proliferation, and apoptosis (26). Further, multiple pro-inflammatory mediators are downstream of IFN-I signaling; thus, IFN-I can regulate multiple physiological processes. Despite discovery of IFN-I over 50 years ago (27), its mechanisms of action with respect to immune modulation (25) or antiviral activity (28, 29) remain unsettled.
Chronic immune activation after HIV infection is documented, and suppression of this hyperactivated state may alleviate pathologies associated with HIV infection (7). Disease after experimental simian immunodeficiency virus (SIV) infection in rhesus macaques correlates with elevated IFN-I and inflammatory signatures (30, 31). In contrast, SIV infection in sooty mangabeys and African green monkeys, which develop modest pathology despite similar viral loads as macaques, correlates with reduced IFN-I and inflammatory signatures (32).
Similar correlations with respect to reduced immune activation exist in HIV-infected elite controllers, although whether reduced immune activation follows better control of virus infection is debatable (33, 34). Moreover, an elevated interferon signature is observed in HCV-infected patients despite limited control of virus replication and development of liver pathology (35, 36). Thus, the IFN-I signaling pathway may be a viable target to control persistent viral infections.
| |
| |
| |
|
|
|