| |
SR9009 Reversed Effect of Mitochondrial Damage,
Increased Exercise Capacity in Mice
|
| |
| |
Download the PDF here
Nature Medicine research article below, authors say: "In conclusion, Rev-erb-α is a major physiological regulator of mitochondrial content and oxidative function. Thus, pharmacological activation of Rev-erb-α may be a promising approach for the treatment of skeletal muscle diseases with compromised exercise capacity.......Rev-erb-α activation improves muscle mitochondrial function......Next, we determined the effect of pharmacological activation of Rev-erb-α on exercise capacity, which we achieved by treating mice with the synthetic ligand SR9009 (ref. 12). Notably, in an endurance exercise test, mice treated with SR9009 ran significantly longer, both in time and in distance, than mice treated with vehicle"
------------------------------------
Drug SR9009 Mimics Exercise Boosting Metabolic Activity And Running Capacity
The Huffington Post Canada | Posted: 08/26/2013
Not in the mood for the gym today?
Or, maybe ever?
It's all right. There may soon be a pill for that.
Scientists say experimental drug SR9009 may boost metabolic activity and running capacity -- essentially mimicking the effects of exercise.
The findings, published in Nature Medicine last month, may be just what the doctor ordered for people suffering from chronic obesity, congestive heart failure -- or any condition which makes physical exercise difficult.
So far, of course, the drug, which was originally developed in 2012, has only been tested on mice. But with the drug increasing running capacity 50-fold in mice, Nature World News reports, can supermen be far behind?
"The animals actually get muscles like an athlete who has been training," said Thomas Burris, a professor at The Scripps Research Institute in Florida, who originally developed the compound. "The pattern of gene expression after treatment with SR9009 is that of an oxidative-type muscle- again, just like an athlete."
The latest study was conducted by an international team of scientists at Institut Pasteur de Lille in France and focused on a naturally occurring molecule in the human body called Rev-erbα.
Researchers say SR9009 latches onto Rev-erbα -- which plays a role in regulating the body's circadian rhythm and metabolism -- resulting in a pronounced metabolic boost in test subjects.
----------------------------------
New Drug Candidate Improves Endurance
Jul 15, 2013.......http://www.natureworldnews.com
A new drug designed by researchers at the Florida campus of The Scripps Research Institute (TSRI) can raise endurance levels in animal test models. The drug, if proven to work on humans, can help people who suffer from obesity and at a risk of heart failure, exercise without any concerns.
The new drug compound called SR9009 is part of a pair of drug candidates developed by the researchers. The drugs work by altering the biological clock of the body. They especially affect a molecule called Rev-erbα. The molecule is highly active in the liver, skeletal muscle, adipose tissue and the brain where it regulates the circadian rhythm of the tissues.
In the study, researchers found that, the drug increased the running capacity by 50 times. The drug activated Rev-erbα in the animals and led to increased metabolic activity in the skeletal muscles. According to the researchers, the drug works by increasing the production of mitochondria (power house) in the cells and also removing the defective mitochondria.
"The animals actually get muscles like an athlete who has been training. The pattern of gene expression after treatment with SR9009 is that of an oxidative-type muscle- again, just like an athlete," Thomas Burris from The Scripps Research Institute, said in a press release.
The study was published July 14, 2013, by the journal Nature Medicine. (FULL TEXT STUDY BELOW)
------------------------------------------
Drug Candidate Leads to Improved Endurance
http://www.scripps.edu/newsandviews/e_20130729/burris.html
By Eric Sauter
An international group of scientists has shown that a drug candidate designed by scientists from the Florida campus of The Scripps Research Institute (TSRI) significantly increases exercise endurance in animal models.
These findings could lead to new approaches to helping people with conditions that acutely limit exercise tolerance, such as obesity, chronic obstructive pulmonary disease (COPD) and congestive heart failure, as well as the decline of muscle capacity associated with aging.
The study was published July 14, 2013, by the journal Nature Medicine.
The drug candidate, SR9009, is one of a pair of compounds developed in the laboratory of TSRI Professor Thomas Burris and described in a March 2012 issue of the journal Nature as reducing obesity in animal models. The compounds affect the core biological clock, which synchronizes the rhythm of the body's activity with the 24-hour cycle of day and night.
The compounds work by binding to one of the body's natural molecules called Rev-erbα, which influences lipid and glucose metabolism in the liver, the production of fat-storing cells and the response of macrophages (cells that remove dying or dead cells) during inflammation.
http://www.healthline.com: SR9009 is one of a pair of compounds developed at TSRI by Burris and his team. The drug binds to Rev-erbα, a natural protein in the body that influences lipid and glucose metabolism in the liver, inflammatory responses, and fat-storing cells. By binding to Rev-erbα, SR9009 can kickstart metabolism with another pleasant side-effect: increased muscle strength. "It 'transforms' muscle into muscle that by many attributes appears to be exercising," Burris explains.
In the new study, a team led by scientists at the Institut Pasteur de Lille in France demonstrated that mice lacking Rev-erbα had decreased skeletal muscle metabolic activity and running capacity. Burris' group showed that activation of Rev-erbα with SR9009 led to increased metabolic activity in skeletal muscle in both culture and in mice. The treated mice had a 50 percent increase in running capacity, measured by both time and distance.
"The animals actually get muscles like an athlete who has been training," said Burris. "The pattern of gene expression after treatment with SR9009 is that of an oxidative-type muscle- again, just like an athlete."
The authors of the new study suggest that Rev-erbα affects muscle cells by promoting both the creation of new mitochondria (often referred to as the "power plants" of the cell) and the clearance of those mitochondria that are defective.
http://www.healthline.com:
Coming to a Pharmacy Near You?
"We were the first to develop drugs that target Rev-erbα that could be used in animals, and our first observations were that the animals increased their metabolic rate," Burris says. Burris and his team have been working on Rev-erbα since 2005 and were aware that it plays some role in the regulation of metabolism.
Now, after modulating Rev-erbα's activity with drugs, studies have shown its direct effect on skeletal muscles.
With such strong results, Burris and his team are pursuing funding for a biotechnology company that would focus primarily on the development of SR9009 and other similar compounds for safe use in humans.
In the next year or so, they hope to start phase I clinical trials. Just imagine, weight loss and improved musculature in a bottle.
---------------------------
Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy
Nature Medicine
Published online 14 July 2013
Estelle Woldt1-5,9, Yasmine Sebti1-5,9, Laura A Solt6, Christian Duhem1-5, Steve Lancel4,7, Jerome Eeckhoute1-5, Matthijs K C Hesselink8, Charlotte Paquet1-5, Stephane Delhaye1-5, Youseung Shin6, Theodore M Kamenecka6, Gert Schaart8, Philippe Lefebvre1-5, Remi Neviere4,7, Thomas P Burris6, Patrick Schrauwen8, Bart Staels1-5 & Helene Duez1-5
1Institut Pasteur de Lille, Lille, France. 2Institut National de la Sante et de la Recherche Medicale Unite Mixte de Recherche 1011 'Nuclear Receptors, Cardiovascular Diseases and Diabetes', Lille, France. 3Faculte des Sciences Pharmaceutiques et Biologiques et Faculte de Medecine, Universite Lille Nord de France, Lille, France. 4Universite du Droit et de la Sante de Lille, Lille, France. 5European Genomic Institute for Diabetes, Lille, France. 6Department of Molecular Therapeutics, The Scripps Research Institute, Jupiter, Florida, USA. 7Departement de Physiologie Equipe d'Accueil 4484, Faculte de Medecine, Universite Lille Nord de France, Lille, France. 8School for Nutrition, Toxicology and Metabolism, Department of Human Biology and Department of Human Movement Sciences, Maastricht University Medical Center, Maastricht, The Netherlands.
"In conclusion, Rev-erb-α is a major physiological regulator of mitochondrial content and oxidative function. Thus, pharmacological activation of Rev-erb-α may be a promising approach for the treatment of skeletal muscle diseases with compromised exercise capacity.......Rev-erb-α activation improves muscle mitochondrial function......Next, we determined the effect of pharmacological activation of Rev-erb-α on exercise capacity, which we achieved by treating mice with the synthetic ligand SR9009 (ref. 12). Notably, in an endurance exercise test, mice treated with SR9009 ran significantly longer, both in time and in distance, than mice treated with vehicle"
"Together, these results demonstrate that Rev-erb-α regulates muscle cell mitochondrial content and function.......Mitochondrial oxidative capacity was impaired in muscle from Nr1d1-/- mice.......This was further underscored by the significantly lower respiration rate observed when comparing equal amounts of isolated mitochondria (Fig. 2e), illustrating not only less mitochondrial content but also lower respiratory chain function of isolated mitochondria from Nr1d1-/- compared to WT mice.....These results suggest that Rev-erb-α deficiency results in an increased autophagy flux, thus contributing to the lower mitochondria number......we demonstrate that autophagy blockade by autophagy or lysosome inhibitors reverses the effect of Nr1d1 knockdown on total mitochondria number.......Autophagy is also a survival mechanism by which cells produce energy in times of nutrient paucity.....Autophagy is a dynamic self-digestion process that ensures the selective clearance of damaged organelles or aggregated macromolecules, which is a particularly important mechanism in differentiated cells such as myotubes26, 27, 28. Our data demonstrate that Rev-erb-α exerts a tight control on this pathway by regulating several genes involved in vesicle nucleation and expansion, autophagosome formation and lysosomal enzymatic activities"
Abstract
The nuclear receptor Rev-erb-α modulates hepatic lipid and glucose metabolism, adipogenesis and the inflammatory response in macrophages. We show here that Rev-erb-α is highly expressed in oxidative skeletal muscle and that its deficiency in muscle leads to reduced mitochondrial content and oxidative function, as well as upregulation of autophagy. These cellular effects resulted in both impaired mitochondrial biogenesis and increased clearance of this organelle, leading to compromised exercise capacity. On a molecular level, Rev-erb-α deficiency resulted in deactivation of the Lkb1-Ampk-Sirt1-Ppargc-1α signaling pathway. These effects were recapitulated in isolated fibers and in muscle cells after knockdown of the gene encoding Rev-erb-α, Nr1d1. In complementary experiments, Rev-erb-α overexpression in vitro increased the number of mitochondria and improved respiratory capacity, whereas muscle overexpression or pharmacological activation of Rev-erb-α in vivo increased exercise capacity. This study identifies Rev-erb-α as a pharmacological target that improves muscle oxidative function by modulating gene networks controlling mitochondrial number and function.
DISCUSSION
Our results from both gain- and loss-of-function experiments identify Rev-erb-α as a physiological regulator of muscle mitochondrial content and oxidative function. This is supported by several observations. First, Rev-erb-α is preferentially expressed in more oxidative muscles, such as soleus muscle, and Nr1d1-/- mice have notably altered exercise capacity associated with a marked decrease in mitochondrial content and function, the presence of swollen mitochondria and abundant vacuoles within the fibers. We observed similar alterations upon knockdown of Nr1d1 in differentiated C2C12 myotubes (lower mitochondrial content and impaired mitochondrial function). By contrast, pharmacological activation or skeletal muscle-specific Rev-erb-α overexpression in vivo and in vitro in C2C12 cells resulted in the opposite phenotype, highlighting the fact that Rev-erb-α exerts a direct action on skeletal muscle cells. Second, our data indicate that Rev-erb-α controls mitochondrial biogenesis and respiration through the Lkb1-Ampk-Sirt1-Ppargc-1α signaling pathway. Finally, we show that skeletal muscle Rev-erb-α regulates several genes involved in different steps of the autophagy process, including genes more specifically dedicated to mitophagy. Together, these data support the concept that Rev-erb-α acts through a two-pronged mechanism, involving both biogenesis of new mitochondria and clearance of defective mitochondria. Thus, when Rev-erb-α is activated, there is an increase in mitochondrial number and a better control of autophagic flux, allowing for a higher oxidative capacity.
The regulation of the Lkb1-Ampk-Sirt1-Ppargc-1α pathway by Rev-erb-α provides a molecular mechanism whereby Rev-erb-α controls mitochondrial biogenesis and function. Ampk and Sirt1 are crucial in the metabolic flexibility that allows skeletal muscle to switch to lipid oxidation during fasting and exercise. Deficient Ampk activity in Nr1d1-/- mice is associated with impaired Sirt1-mediated Ppargc-1α deacetylation and the ensuing attenuated induction of mitochondrial fatty acid oxidation gene expression and reduced exercise capacity20, 23. Likewise, Ppargc-1α acetylation is higher and its expression diminishes in Rev-erb-α deficiency, which may contribute to the reduced number of mitochondria, lower expression of transcription factors important for mitochondrial function, such as Tfam, and altered electron transport chain activity. In addition, expression of genes encoding fatty acid ß-oxidation enzymes as well as palmitate-induced mitochondrial respiration is compromised in the absence of Rev-erb-α. A considerable deactivation of the Lkb1-Ampk-Sirt1-Ppargc-1α pathway when Rev-erb-α is deficient is further shown by defective Ampk activation, reduced NAD+ concentrations and markedly altered Sirt1 and Nampt expression. Moreover, siRNA-mediated or C compound-mediated inhibition of Ampk activity, which blocks the stimulatory effect of REV-ERB-α overexpression on mitochondrial respiration, demonstrates that Rev-erb-α-mediated improvement of mitochondrial respiration requires active Ampk. Of note, in Nr1d1-/- mice, Ampk phosphorylation is not induced even though ATP concentrations are low. Binding of AMP increases the ability of other kinases to phosphorylate and activate the Ampk-α subunit. Lkb1, expressed in skeletal muscle, is a major upstream kinase that phosphorylates and activates Ampk, and Stk11-deficiency results in blunted Ampk activation, decreased Ppargc1a expression, reduced exercise capacity and compromised mitochondrial activity24, 25, a phenotype similar to the one observed in the Nr1d1-/- mice. Our results also indicate that mitochondria are damaged and mitochondrial oxygen consumption is reduced in muscle from Nr1d1-/- mice, whereas skeletal muscle-specific Rev-erb-α overexpression improves mitochondrial respiration. Moreover, treatment of mice with a synthetic Rev-erb-α agonist increases oxygen consumption12. Notably, and in contrast to other transcriptional regulators, such as Ppar-ß/δ (ref. 2) and Ppargc-1α (refs. 1,6), Rev-erb-αmodulates skeletal muscle oxidative capacity without inducing a fiber type switch, suggesting a disconnection between both phenomena upon Rev-erb-α deficiency. Together, these data support a prominent role for Rev-erb-α in skeletal muscle mitochondria function.
Autophagy is a dynamic self-digestion process that ensures the selective clearance of damaged organelles or aggregated macromolecules, which is a particularly important mechanism in differentiated cells such as myotubes26, 27, 28. Our data demonstrate that Rev-erb-α exerts a tight control on this pathway by regulating several genes involved in vesicle nucleation and expansion, autophagosome formation and lysosomal enzymatic activities. The conversion of Map1lc3a-I to its lipidated form, Map1lc3a-II, is a hallmark of autophagy29. The rate of maturation to Map1lc3a-II was elevated in the absence of Rev-erb-α, whereas autophagy flux is decreased by REV-ERBα overexpression. Of note, Rev-erb-α represses genes encoding proteins that trigger mitophagy, such as Park2, a cytosolic E3 ubiquitin ligase that translocates to depolarized mitochondria to induce mitophagy, thereby maintaining a pool of functioning mitochondria and limiting oxidative damage21. Likewise, Rev-erb-α downregulates Ulk1, which not only participates in the initiation complex but also may play an important part in triggering mitophagy30. Ulk1 deficiency indeed results in accumulation of defective mitochondria in mature red blood cells31. Finally, we demonstrate that autophagy blockade by autophagy or lysosome inhibitors reverses the effect of Nr1d1 knockdown on total mitochondria number.
Autophagy is also a survival mechanism by which cells produce energy in times of nutrient paucity. One of the most potent inducers of autophagy is Ampk, which directly phosphorylates Ulk1 and, simultaneously, turns off signaling of mTORC1, an inhibitor of autophagy26, 28, 32. However, neither Ampk (which is downregulated in the absence of Rev-erb-α) nor mTOR, whose signaling is not regulated by Rev-erb-α (data not shown), is likely to mediate Rev-erb-α's action on autophagy. Another potent autophagy inducer, Sirt1 (ref. 33), is also downregulated in the absence of Rev-erb-α, again excluding the possibility that Sirt1 has a role in the observed phenotype. Together, these data indicate that Rev-erb-α bypasses these regulators and represses autophagy genes directly, probably by binding the DNA in their regulatory regions. Indeed, our ChIP-qPCR data confirmed Rev-erb-α binding enrichment in the regulatory regions of autophagy genes. This was accompanied by a modification of epigenetic marks at these sites, as indicated by decreased H3K27 and H3K9 acetylation, which is consistent with the repressive role of Rev-erb-α on gene transcription.
Rev-erb-α is a component of the circadian clock, which allows synchronization of internal rhythms to daily environmental cues34. Skeletal muscle has circadian rhythmicity of gene expression35, 36, and the clock components Clock and Bmal1 (brain and muscle Arnt-like 1) have been shown to participate in the maintenance of skeletal muscle function37. The mRNA concentrations of the clock genes are altered in skeletal muscle from Nr1d1-/- mice (Supplementary Fig. 7a), and REV-ERB-α overexpression in synchronized C2C12 cells leads to an altered circadian expression pattern of Ppargc1a (Supplementary Fig. 7b).
Pharmacological activation of Rev-erb-α alters the circadian rhythms of Cpt1b and Ppargc1a in skeletal muscle12. Consequently, in addition to the direct action of Rev-erb-α on autophagy genes and mitochondrial biogenesis and function, Nr1d1 deletion or overexpression that causes disturbed circadian rhythmicity may contribute to the observed phenotype. In conclusion, Rev-erb-α is a major physiological regulator of mitochondrial content and oxidative function. Thus, pharmacological activation of Rev-erb-α may be a promising approach for the treatment of skeletal muscle diseases with compromised exercise capacity.
Introduction
Skeletal muscle contractility is important for locomotion and posture, activities performed by different myofiber types with distinct contractile and metabolic properties. Mitochondria serve a crucial function in the maintenance of skeletal myofiber homeostasis and matching energy production with demand. They do so through oxidation of glucose-derived pyruvate and ß-oxidation of fatty acids, generating an electrochemical proton gradient through the respiratory complexes of the electron transport chain; this gradient can be used to drive the phosphorylation of ADP to ATP, a reaction called oxidative phosphorylation (OXPHOS). However, what determines mitochondrial content and function is not completely understood.
Nuclear receptors and their cofactors regulate metabolism in response to environmental signals and trigger homeostatic responses by coordinately regulating transcriptional networks. Previous studies have established that nuclear receptors, such as peroxisome proliferator-activated receptor-ß/δ (Ppar-ß/δ), estrogen-related receptor-α and estrogen-related receptor-γ, along with co-regulators such as Ppar-γ coactivator-1α (Ppargc-1α) and Ppargc-1ß, as well as nuclear receptor co-repressor 1 (Ncor1), among others, control muscle physiology by modulating mitochondrial biogenesis and function, fiber type determination and switching and muscle vascularization1, 2, 3, 4, 5. P pargc-1α is a master driver of mitochondrial biogenesis, and its overexpression in skeletal muscle results in increased mitochondrial number and function1, whereas skeletal muscle Ppargc-1α deficiency leads to a reduced number of mitochondria and a marked reduction of muscle oxidative capacity6, 7.
The nuclear receptor Rev-erb-α is expressed in tissues such as liver and adipose tissue8, where it modulates lipid, bile acid and glucose metabolism8, 9, 10, 11, 12, 13. In addition, Rev-erb-α controls adipogenesis14, 15 and the macrophage inflammatory response16. Rev-erb-α interacts with Ncor1 and chromatin modifiers, such as histone deacetylase 3, to form a complex repressing target gene transcription17. Notably, a Rev-erb-α-Ppargc-1α cross-talk pathway regulates heme synthesis in hepatic cells18, 19. Whether Rev-erb-α interacts with Ppargc-1α in muscle and whether Rev-erb-α controls skeletal muscle oxidative capacity has not yet been investigated. Here we find through loss- and gain-of-function experiments, including pharmacological activation, that Rev-erb-α plays a key role in regulating the oxidative capacity of the muscle and exercise endurance, and thus it emerges as a potential target to improve muscle function.
RESULTS
Nr1d1-/- mice have lower exercise capacity
Rev-erb-α expression is markedly higher in oxidative compared to more glycolytic muscles (Fig. 1a). Notably, its expression is higher in soleus and gastrocnemius muscle upon exercise training (Supplementary Fig. 1a). Thus, we explored whether Rev-erb-α has a role in skeletal muscle oxidative capacity and exercise capacity. Nr1d1-/- mice showed significantly less spontaneous locomotor activity in a free-wheel exercise regimen compared to wild-type (WT) littermates (Fig. 1b).
We next assessed basal (VO2b) and maximal (VO2max) oxygen consumption, which reflect aerobic capacity, by submitting Nr1d1-/- and WT mice to a forced progressive treadmill exercise. VO2b measured at rest was not significantly different between the two genotypes, whereas VO2max measured at exhaustion was significantly lower in Nr1d1-/- mice (Fig. 1c), resulting in a >60% reduction in aerobic capacity during exercise. In a standard endurance exercise test performed at 70% of their respective VO2max, 50% of Nr1d1-/- mice, compared to only 20% of the WT mice, stopped running within 50 min, which indicates their inability to sustain a long-lasting exercise (Fig. 1d). In this setting, Nr1d1-/- mice ran for a significantly shorter time and distance than their WT littermates (Fig. 1e,f).
Rev-erb-α controls muscle mitochondrial content and function
We assessed the role of Rev-erb-α in the control of mitochondrial number and function in vivo (Fig. 2). Mitochondrial DNA content was ~40% lower in skeletal muscle from Nr1d1-/- mice compared to WT littermates (Fig. 2a), suggesting that Rev-erb-α is involved in regulating skeletal muscle mitochondrial content. Accordingly, expression of genes encoding subunits of the mitochondrial electron transport respiratory chain, such as NADH dehydrogenase 1, a subunit of complex I, and cytochrome c oxidase 1 and cytochrome c oxidase 2, two subunits of complex IV, was lower in soleus and quadriceps muscle from Nr1d1-/- mice compared to WT mice (Fig. 2b and Supplementary Fig. 1b). In addition, relative protein amounts of the mitochondrial oxidative phosphorylation complexes were lower in muscle from Nr1d1-/- mice compared to WT mice (Fig. 2c and Supplementary Fig. 1c). Moreover, glutamate-malate-stimulated (state 2) respiration was lower in isolated fibers from Nr1d1-/- mice compared to WT mice, in line with the reduced mitochondrial content (Fig. 2d). Mitochondrial oxidative capacity was impaired in muscle from Nr1d1-/- mice, as attested by a significantly lower ADP-driven glutamate-malate (state 3) respiration rate in saponin-permeabilized fibers isolated from Nr1d1-/- compared to WT mice (Fig. 2d). The lower respiration upon addition of succinate and rotenone demonstrates a reduced capacity of the entire chain rather than a deficiency in a specific complex. This was further underscored by the significantly lower respiration rate observed when comparing equal amounts of isolated mitochondria (Fig. 2e), illustrating not only less mitochondrial content but also lower respiratory chain function of isolated mitochondria from Nr1d1-/- compared to WT mice.
Although gene expression analysis of fiber type markers suggested a switch toward a more glycolytic profile (expression of the genes encoding tropomyosin 3, a marker of oxidative type I fiber, and myosin heavy chain IIa and IIx, markers of mostly oxidative fibers, is lower in soleus and quadriceps muscle from Nr1d1-/- mice compared to WT mice (data not shown)), specific type I, type IIa and type IIb immunostaining did not reveal any significant changes between the two genotypes (Supplementary Fig. 1d). In addition, expression of Mb (the gene encoding myoglobin) and vascularization, assessed by CD31 immunostaining on soleus muscle sections, was similar between the two genotypes (Supplementary Fig. 5d,e).
To study whether Rev-erb-α regulates mitochondrial function in a cell-autonomous manner, we stably infected C2C12 muscle cells with a REV-ERB-α-coding retrovirus. Staining with MitoTracker Green, a marker of mitochondria content, was higher in cells overexpressing REV-ERB-α compared with those infected with a control vector (Fig. 3a). Notably, the red-to-green ratio of JC-1 fluorochrome, an indicator of membrane potential, was also higher in C2C12 cells overexpressing REV-ERB-α compared to cells infected with an empty pBabe control retrovirus, suggesting increased mitochondrial activity (Fig. 3b). REV-ERB-α expression enhanced the maximal respiratory capacity through enhanced coupled ATP-producing oxidative phosphorylation, as illustrated by higher-state 3u respiration rate and respiratory control ratio (RCR, OXPHOS/leak), whereas the uncoupling proton leakage remained unchanged (Fig. 3c). Conversely, Nr1d1 silencing resulted in lower mitochondrial respiration compared to control shRNA-infected cells (Supplementary Fig. 2a), as well as the quantity of total (MitoTracker Green) and functional (MitoTracker Red) mitochondria (Supplementary Fig. 2b) in differentiated C2C12 cells. Analysis of mitochondrial fatty acid oxidation in permeabilized fibers of muscle from Nr1d1-/- mice revealed compromised oxygen consumption in the presence of palmitoyl-L-carnitine plus malate alone and in the presence of ADP (Fig. 2f). In parallel, the expression of genes encoding enzymes of fatty acid ß-oxidation, notably carnitine palmitoyltransferase 1B (Cpt1b) and (very) long chain (Acadvl and Acadl) and short chain (Acads) acyl-CoA dehydrogenases, was lower in skeletal muscle from Nr1d1-/- compared with WT mice (Fig. 2g and Supplementary Fig. 3a), whereas REV-ERB-α retrovirus-infected C2C12 cells had a mirror phenotype (that is, higher Acadvl, Acadl and Acadsexpression, and thus a higher acyl-CoA dehydrogenase expression) compared to control cells infected with an empty control pBabe retrovirus (Fig. 3d). These results indicate that Rev-erb-α modulates fatty acid ß-oxidation-driven generation of reducing equivalents to feed into the electron transport chain in vitro and ex vivo in skeletal muscle. Together, these results demonstrate that Rev-erb-α regulates muscle cell mitochondrial content and function.
Electron microscopy analysis of muscle sections revealed a slight misalignment of Z lines, the presence of vacuolated fibers and the presence of abnormal, swollen and less dense mitochondria in muscle sections from Nr1d1-/- mice compared to WT littermates (Fig. 2h and Supplementary Fig. 4), which indicate a severe skeletal muscle phenotype and mitochondrial dysfunction in the Nr1d1-/- mice. The repair process, assessed by the presence of centrocellular nuclei (Supplementary Fig. 5a shows that the nuclei are localized at the periphery of the fibers, confirming the absence of regeneration) and the absence of change in the expression of Pax7 and Myf5 (Supplementary Fig. 5b), was not different between Nr1d1-/- mice and WT littermates. In line with this, immunofluorescence staining of Pax7 indicated its localization at the boundary of the myofiber (Supplementary Fig. 5c), which implies the presence of quiescent satellite cells located on the border of myofibers, which are similar in the two genotypes.
Rev-erb-α increases skeletal muscle mitochondrial biogenesis
We assessed whether Rev-erb-α regulates mitochondrial biogenesis in loss- and gain-of-function settings (Fig. 4). Both mRNA (~56%) and protein (~50%) amounts of Ppargc-1α, which has a pivotal role in mitochondrial biogenesis, were lower in soleus and quadriceps muscle from Nr1d1-/- mice as compared to WT mice (Fig. 4a and Supplementary Fig. 3b,d). Consistently, the expression of Tfam and Nrf1, encoding mitochondrial transcription factor A and nuclear respiratory factor 1, respectively, two transcription factors that are involved in mitochondrial biogenesis, was lower in Nr1d1-/- mice compared to WT mice (Fig. 4b). By contrast, REV-ERB-α overexpression resulted in higher Ppargc1a and Tfam expression in C2C12 cells compared to cells infected with an empty pBabe control retrovirus (Fig. 4h).
As a functional reflection of impaired mitochondrial electron transport chain activity, ATP concentrations were significantly lower in muscle from Nr1d1-/- mice compared to WT mice (Fig. 4c). Ampk, a 'fuel gauge' activated by liver kinase B1 (Lkb1, also known as serine-threonine kinase 11 or Stk11) when the AMP/ATP ratio increases, induces Sirt1-dependent deacetylation of Ppargc-1α and expression of nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme in the synthesis of the Sirt1 cofactor NAD+, thus affecting mitochondrial and lipid oxidation genes20. Of note, Stk11 gene expression (Fig. 4d and Supplementary Fig. 3d), Ampk phosphorylation (Fig. 4e) and activity illustrated by the p-Acac/Acac (phosphorylated acetyl-CoA carboxylase/total acetyl-CoA carboxylase) ratio (Supplementary Fig. 3c) and Nampt gene expression (Fig. 4f) were lower in muscle from Nr1d1-/- mice compared to WT mice. Concentrations of NAD+ and NADH, the reducing equivalent produced by fatty acid oxidation, were lower in skeletal muscle from Nr1d1-/- mice compared to that of their WT littermates (Fig. 4f). Moreover, we observed significantly less Sirt1 expression in skeletal muscle from Nr1d1-/- compared to WT mice (Fig. 4f and Supplementary Fig. 3d), and SIRT1 activity was blunted, as attested by increased Ppargc-1α acetylation (Fig. 4g). Conversely, Sirt1 and Nampt expression was induced in REV-ERB-α-overexpressing C2C12 cells (Fig. 4h) and accompanied by significantly improved mitochondrial respiration, an effect that was fully blocked by the addition of the C compound, an inhibitor of Ampk phosphorylation (Fig. 4i). Consistent with this, siRNA knockdown of genes encoding Ampk (Prkaa1 and Prkaa2) prevented the increase in mitochondrial content upon REV-ERB-α overexpression (Fig. 4j), whereas Ampk activation by 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) increased mitochondrial number in cells stably transduced with Nr1d1 shRNA but not to the same extent as in control cells stably transduced with control shRNA (Supplementary Fig. 2c). Together, these data indicate that Rev-erb-α regulates skeletal muscle mitochondria biogenesis through modulation of the Lkb1-Ampk-Sirt1-Ppargc-1α pathway.
Rev-erb-α deficiency induces skeletal muscle autophagy
We next explored whether Rev-erb-α also modulates mitochondrial degradation. Autophagy is a process mediating the selective clearance of cytoplasmic components, such as damaged mitochondria, that could otherwise become deleterious. Autophagy is a self-digestion process occurring through the formation of a vesicle (nucleation) that expands to become an autophagosome, which then engulfs cellular components and directs them to the lysosome for degradation. Expression of the genes encoding Ulk1, a protein of the initiation complex implicated in vesicle formation, and Beclin1, a protein of the nucleation complex, as well as autophagy-related 5 (Atg5) and Bnip3, which are responsible for vesicle elongation and autophagosome formation, and the lysosomal enzymes cathepsin L (Ctsl) and ATPase6v1b2 was higher in skeletal muscle of Nr1d1-/- compared to WT mice (Fig. 5a,b) and lower in REV-ERB-α retrovirus-infected C2C12 cells compared to control cells infected with an empty pBabe control retrovirus (Supplementary Fig. 6). Rev-erb-α also regulated gene and protein expression of Parkin (also known as Park2), a protein specifically involved in mitochondrial clearance21 (Fig. 5b and Supplementary Fig. 6). The functional increase of autophagy was illustrated by the maturation of microtubule-associated protein 1 light chain 3α (Map1lc3a-I, also known as LC3) to its lipidated form, Map1lc3a-II, a marker of ongoing autophagy associated with the autophagosome membrane (Fig. 5c). Moreover, treatment with the lysosome inhibitors bafilomycin and NH4Cl, which block the fusion of the lysosome with mature autophagosomes that subsequently accumulate, resulted in a smaller increase of Map1lc3a-II expression in REV-ERB-α-overexpressing C2C12 cells than in treated control (pBabe) cells (Fig. 5d). This indicates that autophagy flux is lower when REV-ERB-α is overexpressed. Conversely, the number of mitochondria was lower after Nr1d1 silencing in C2C12 cells, and addition of the autophagy blockers 3-methyl adenine and chloroquine or the lysosome inhibitors bafilomycin and NH4Cl prevented this decrease (Fig. 5e). These results suggest that Rev-erb-α deficiency results in an increased autophagy flux, thus contributing to the lower mitochondria number.
To gain further mechanistic insight, we examined the distribution of Rev-erb-α binding sites in the vicinity of autophagy genes, assessed using chromatin immunoprecipitation (ChIP) sequencing data17, and overlaid it with epigenetic marks obtained in C2C12 myotubes22. We reasoned that intense transcriptional activity at these marks may increase the likelihood of Rev-erb-α binding to these locations. Primers for ChIP-quantitative PCR (qPCR) targeting these regions were then designed to assess REV-ERB-α binding and changes in acetylation marks in REV-ERB-α-expressing C2C12 cells. We observed an enrichment of Rev-erb-α binding in the regulatory regions of several autophagy genes (Fig. 5f), which was associated with decreased acetylation at histone 3 Lys27 (H3K27) (Fig. 5g) and Lys9 (H3K9) (Fig. 5h).
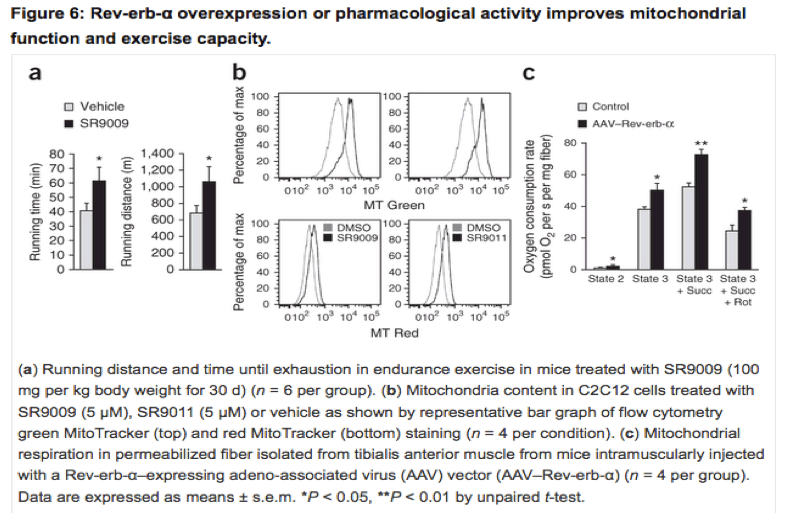
Rev-erb-α activation improves muscle mitochondrial function
Next, we determined the effect of pharmacological activation of Rev-erb-α on exercise capacity, which we achieved by treating mice with the synthetic ligand SR9009 (ref. 12). Notably, in an endurance exercise test, mice treated with SR9009 ran significantly longer, both in time and in distance, than mice treated with vehicle (Fig. 6a).
Incubation of C2C12 cells with two different Rev-erb agonists, SR9009 and SR9011, increased the number of total (MitoTracker Green) and active (MitoTracker Red) mitochondria (Fig. 6b). Finally, skeletal muscle-specific Rev-erb-α overexpression in mice significantly improved mitochondrial function, as attested by a significant increase in glutamate-malate-stimulated and ADP-driven respiration in the absence and presence of succinate in permeabilized fibers (Fig. 6c). Together, these data indicate that enhancement of Rev-erb-α expression, activity or both exerts beneficial direct effects on skeletal muscle to improve mitochondrial respiration and exercise capacity.
|
|
| |
| |
|
|
|