| |
HIV Protein Causes Metabolic/Fat Abnormalities, New Study: HIV-1 Vpr Induces Adipose Dysfunction in Vivo Through Reciprocal Effects on PPAR/GR Co-Regulation
|
| |
| |
Download the PDF here
Download the PDF here
- full text below
Sci Transl Med 27 November 2013
"Viral infections, such as HIV, have been linked to obesity, but mechanistic evidence that they cause adipose dysfunction in vivo is lacking......Vpr circulated in the blood of most HIV-infected patients tested, including those on antiretroviral therapy (ART) with undetectable viral load.
........Vpr-mediated mechanisms were dissected in vivo using mouse models expressing the Vpr transgene in adipose tissues and liver (Vpr-Tg) or infused with synthetic Vpr........Vpr (HIV viral protein) circulates in the blood of HIV patients even after viral-suppressive treatment with ART, and its presence in the circulation of mice is sufficient to recapitulate the characteristic features of adipose and hepatic metabolic defects observed in HIV patients: accelerated lipolysis (17-19), diminished fat mass (7, 28), hepatosteatosis (20), insulin resistance, and hyperglycemia (29)...............Together with in vitro evidence for cellular- and nuclear-transducing activities of virion-free Vpr (16), these data demonstrate that Vpr drives mechanisms that may underlie the metabolic defects in HIV patients. Vpr, produced by infected immune cells within reservoirs in HIV patients on ART (15), can transduce adipose tissues and liver to produce these metabolic defects........The current data are limited in demonstrating the critical Vpr mechanisms in mice-full translation would require longitudinal metabolic measurements in blood and tissue specimens of HIV patients on cART with a range of serum Vpr levels and VL. In a cross-sectional analysis, we found no correlation between serum Vpr levels and lipid kinetic rates in 12 hyperlipolytic, hypertriglyceridemic HIV patients on cART with a range of VL, suggesting that Vpr has adipose-restricted effects not reflected quantitatively in serum Vpr concentrations.......We infer that Vpr in the serum of patients reflects its production by HIV sequestered in adipose tissue, liver, or other reservoirs but we have not measured Vpr production relative to HIV replication within those tissues or the kinetics of its extracellular transfer. Vpr is likely not the sole pathogenic factor in HIV-associated metabolic disturbances, but it has an independent effect. Confirmation of these mechanisms in patients could pave the way for targeted treatment with small-molecule inhibitors of Vpr, GR antagonists, or dual PPARγ/PPARα agonists."
---------------------------------------------
press announcement from the Baylor College of Medicine, full published study follows below-
HIV-1 accessory protein helps induce metabolic defects
The wily human immunodeficiency virus (HIV-1) has another trick up its sleeve, one that can disrupt cellular metabolism and raise the risk of heart disease, diabetes and osteoporosis in patients already receiving treatment to fight HIV, said researchers led by those at Baylor College of Medicine in a report that appears online in the journal Science Translational Medicine.
The culprit is an HIV-1 accessory protein known as viral protein R (Vpr). The findings reported by the researchers answer some of the questions as to why patients receiving successful treatment for HIV end up with abnormal fat loss and deposits - a problem call lipodystrophy.
"With antiretroviral treatment, HIV has become a chronic illness, and lipodystrophy and associated metabolic defects have become, long-term, serious complications," said Dr. Ashok Balasubramanyam, professor of medicine - diabetes, endocrinology and metabolism at BCM. "It appears that latent or hidden HIV is dangerous not only because it can flare up if treatment is interrupted, but also because it keeps producing toxic signals that make HIV disease (despite the best antiviral treatment) a chronic, metabolically debilitating illness with high risk of diabetes, cardiovascular disease and obesity. This adds to the urgent need for a real cure for HIV, i.e., ways of wiping out all traces of HIV even in reservoirs where it currently hides out."
Because the HIV lipodystrophy syndrome was first noticed around the time that the successes of anti-retroviral drugs became apparent, most clinicians assumed that the drugs were the cause. However, research shows that while the drugs may play a role, some of the problems with lipids in the blood and glucose levels and diseases associated with those problems continue, even if the drugs are changed or newer drugs are used. Fat metabolic defects also exist in patients who are untreated or have a genetic quirk that prevents the virus from destroying their immune systems after infection.
Focus on HIV01 virus protein R
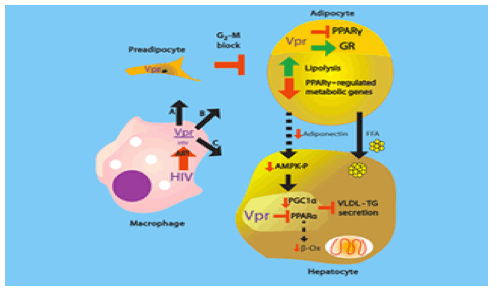
The figure shows how Vpr causes metabolic defects in HIV infected patients. HIV persists in immune cells within certain "reservoirs" in the body even after treatment with HIV drugs.
In this case, Balasubramanyam and his colleagues focused on HIV-1 virus protein R (Vpr), which can co-activate the glucocorticoid receptor and co-repress proliferator-activated receptor gamma (PPARgamma), both factors that play key roles in cellular metabolism.
"We found that Vpr circulates in the blood of patients with no detectable viral load after treatment with the best antiviral drugs," said Balasubramanyam. "This implies that 'latent' or 'persistent' HIV that is hiding out in reservoirs is constantly turning out Vpr, which can cause the metabolic defects."
"It appears to work as a toxic hormone that is sent out from infected cells to attack others than cannot be infected directly by HIV-1," he said. This protein can easily infiltrate fat and liver cells, where it disrupts some of the genetic machinery and regulating proteins crucial to metabolic function.
"These metabolic pathways get disrupted, resulting in excessive breakdown of fat stores, increased release of fats into the bloodstream, and insulin resistance," he said.
In studies in mice, he and his colleagues, including first authors Dr. Neeti Agarwal, a post-doctoral associate, and Dr. Dinakar Iyer, an instructor in the department of medicine - diabetes, endocrinology and metabolism, both at BCM, found that mice that produce Vpr in the liver and fat cells suffer disruption of metabolic pathways. In another experiment, they pumped Vpr in mice as though it were a drug to recreate what happens in patients. Again, the metabolic pathways were disturbed.
"One big surprise was how closely the specific defects in each model resembled the defects in the other - proof that not only is Vpr capable of disrupting these metabolic pathways, but can do so when introduced into the bloodstream of the mice. The second was the finding of fatty liver in these mice. Fatty liver is very prevalent in HIV patients, but we weren't expecting it in these mice," said Balasubramanyam. They found that Vpr interferes with pathways involving proper handling of fat by the liver.
Possible uses
Addressing the mechanisms mediated by Vpr might help HIV patients with the complications of insulin resistance, fatty liver, diabetes and heart disease, he said. One possibility might be using a drug or neutralizing antibodies to block the overall effect of Vpr. It might also be possible to use a drug such as mifepristone to block the glucocorticoid receptor, which Vpr activates.
"These findings add to the urgent need for a real cure for HIV, wiping out all traces of the virus, even in reservoirs where it hides," he said.
It also suggests areas for further research with viruses, particularly those that are chronic or hide in the body for long periods, such as herpes, adenovirus or cytomegalovirus (CMV).
Revise thinking
"It would be a good idea to focus research efforts on uncovering more connections between chronic infections and metabolic diseases. Hitherto infectious diseases and metabolic diseases have been considered to be separate and unconnected - I think it's time to revise our thinking about that," he said.
Others who took part in this research include: Rajagopal V. Sekhar, Toni Oplt, Eric D. Buras, Susan L. Samson, Maria C. Rodriguez-Barradas and Farook Jahoor, all of BCM; Terry M. Phillips and Tomoshige Kino, both of the National Institutes of Health; Ulrich Schubert of the University of Erlangen in Germany; Jacob Couturier and Dorothy.E. Lewis, both of the University of Texas Health Science Center at Houston; Jeffrey B. Kopp of the National Institute of Diabetes and Digestive and Kidney Diseases; and Sanjeet G. Patel of the University of California Los Angeles Medical Center in California. Balasubramanyam, Sekhar and Samson are also with Harris Health System's Ben Taub Hospital.
Rodriguez-Barradas is also with the Michael E. DeBakey Veterans Affairs Medical Center. Buras is now with the University of Michigan in Ann Arbor.
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (Grant DK081553) and a Developmental Grant from the Baylor Center for AIDS Research (National Institute of Allergy and Infectious Diseases [Grant P30AI36211]), a Seed award (NIDDK Grant P30DK079638 [Diabetes Research Center at Baylor]), German Research Council (Grants SFB 796 and 643), the NIDDK Intramural Research Program, and the NIH Office of the Director.
----------------------------------------------
Sci Transl Med 27 November 2013
HIV-1 Vpr Induces Adipose Dysfunction in Vivo Through Reciprocal Effects on PPAR/GR Co-Regulation
ABSTRACT: Viral infections, such as HIV, have been linked to obesity, but mechanistic evidence that they cause adipose dysfunction in vivo is lacking. We investigated a pathogenic role for the HIV-1 accessory protein viral protein R (Vpr), which can coactivate the glucocorticoid receptor (GR) and co-repress peroxisome proliferator-activated receptor γ (PPARγ) in vitro, in HIV-associated adipose dysfunction. Vpr circulated in the blood of most HIV-infected patients tested, including those on antiretroviral therapy (ART) with undetectable viral load. Vpr-mediated mechanisms were dissected in vivo using mouse models expressing the Vpr transgene in adipose tissues and liver (Vpr-Tg) or infused with synthetic Vpr. Both models demonstrated accelerated whole-body lipolysis, hyperglycemia and hypertriglyceridemia, and tissue-specific findings. Fat depots in these mice had diminished mass, macrophage infiltration, and blunted PPARγ target gene expression but increased GR target gene expression. In liver, we observed blunted PPARα target gene expression, steatosis with decreased adenosine monophosphate-activated protein kinase activity, and insulin resistance. Similar to human HIV-infected patients, Vpr circulated in the serum of Vpr-Tg mice. Vpr blocked differentiation in preadipocytes through cell cycle arrest, whereas in mature adipocytes, it increased lipolysis with reciprocally altered association of PPARγ and GR with their target promoters. These results delineate a distinct pathogenic sequence: Vpr, released from HIV-1 in tissue reservoirs after ART, can disrupt PPAR/GR co-regulation and cell cycle control to produce adipose dysfunction and hepatosteatosis. Confirmation of these mechanisms in HIV patients could lead to targeted treatment of the metabolic complications with Vpr inhibitors, GR antagonists, or PPARγ/PPARα agonists.
INTRODUCTION
Viral infections are linked to obesity (1) and fatty liver (2), but evidence that they cause adipose dysfunction is correlative (3). In vivo mechanisms whereby viruses induce adipocyte defects in human adipose disorders have not been reported. HIV patients manifest adipose dysfunction characterized by accelerated lipolysis, lipoatrophy in some depots and lipohypertrophy in others, hepatosteatosis, dyslipidemia, insulin resistance, and hyperglycemia. Antiretroviral therapy (ART) drugs have been implicated in some abnormalities (4). However, adverse effects of ART cannot explain key aspects of the phenotype (5); for example, hypertriglyceridemia was noted before the ART era (6), and decreased body fat (7), altered fat distribution (8), and abnormal adipose gene expression (9, 10) occur in untreated patients. Thus, HIV-1 per se could cause adipose dysfunction and associated metabolic defects. In vivo demonstration of these defects and their mechanisms would provide critical proof of a viral etiology for lipodystrophy or obesity.
Viral protein R (Vpr), an HIV-1 accessory protein, functions in virion assembly, preintegration complex translocation, nucleocytoplasmic shuttling, and transcriptional regulation of the HIV-1 long terminal repeat and host genes (11). Three effects, demonstrated in vitro, could be relevant to adipose metabolism: Vpr (i) potentiates glucocorticoid receptor (GR)-mediated transcription via an LQQLL nuclear receptor co-regulator motif (12, 13); (ii) co-represses peroxisome proliferator-activated receptor γ (PPARγ)-mediated transcription (14); and (iii) induces G2-M cell cycle arrest and apoptosis in infected T cells (15). GR coactivation and PPARγ co-repression in adipocytes and hepatocytes could cause hyperlipolysis and insulin resistance, whereas G2-M arrest in preadipocytes could block differentiation, leading to lipoatrophy.
Two challenges to a plausible role for Vpr in adipose and hepatic dysfunction in HIV patients are as follows: (i) HIV-1 does not infect adipocytes or hepatocytes, so how could Vpr enter these cells? (ii) Lipoatrophy, dyslipidemia, and insulin resistance occur in patients receiving ART with undetectable viral load (VL), so what could be the source of Vpr in these patients? Several characteristics of Vpr could overcome these difficulties. Vpr can be released from HIV-infected cells and circulate independently (16). Moreover, Vpr is produced by replication-deficient HIV-1 and even during inhibition of viral replication by protease inhibitors (15), so it could be released from HIV-1 sequestered in tissue reservoirs in ART-treated patients. Finally, Vpr can transduce cells in a receptor- and energy-independent manner and localize in the cytosol, nucleus, and mitochondria (14, 16).
We hypothesized that virion-free Vpr, with the ability to transduce adipose and hepatic cells, persists in the circulation of HIV patients after treatment with "viral-suppressive" ART and is sufficient to produce the HIV-associated metabolic phenotype through PPARγ co-repression, GR coactivation, and cell cycle arrest in adipose and hepatic tissues. We tested these hypotheses by measuring Vpr in the circulation of HIV-infected patients on ART and specifying Vpr-mediated pathogenic mechanisms in two mouse models: transgenic (expressing Vpr in adipose tissues and liver) and pharmacologic (designed to measure the effects of circulating Vpr).
RESULTS
Vpr circulates in the blood of ART-treated HIV patients with undetectable VL
We measured Vpr by immunoaffinity capillary electrophoresis (ICE) in masked serum samples from HIV-negative persons (n = 20) and three HIV-infected groups: (i) ART-naïve (n = 25), (ii) on nucleoside reverse transcriptase inhibitors (NRTIs) only (n = 61), and (iii) on combination ART (cART, n = 70), of whom 25 had undetectable VL. Ninety-six percent of the HIV patients (88% on ART with undetectable VL) had detectable (true-positive) serum Vpr (Fig. 1A). These data indicate that Vpr produced by HIV-1 persisting in reservoirs can be released into the circulation. Serum Vpr ranges overlapped in the HIV-positive groups; the median value was lower in the cART group than in the treatment-naïve group. There was no correlation between Vpr level and VL among untreated or NRTI-only patients. Vpr was identified in adipose tissues and liver obtained at autopsy of two HIV-infected, but not of three HIV-uninfected, persons (table S1).
Vpr expressed in adipose tissues and liver circulates in the blood of transgenic mice (Vpr-Tg)
Vpr mRNA was detected in adipose tissue and liver of Vpr-Tg mice, which express Vpr under the control of the phosphoenolpyruvate carboxykinase (PEPCK) promoter (Fig. 1B). Expression of Vpr was weaker in liver than in adipose tissue, likely because of Vpr's ability to blunt the expression of PEPCK (see below). ICE demonstrated Vpr in the sera of Vpr-Tg mice [194 ± 7 pg/ml (mean ± SE)], showing that Vpr produced in tissues is released into the bloodstream (Fig. 1C). To generate the pharmacologic model, we defined the pharmacokinetics of synthetic Vpr (sVpr) in wild-type mice using intraperitoneal injections. The serum half-life of sVpr was short (6 to 12 hours, fig. S1); hence, a chronic subcutaneous (Alzet) infusion method (delivering sVpr for 14 days) was adopted. Mean serum Vpr concentration in these sVpr-treated mice was 755 ± 16 pg/ml after 14 days (Fig. 1C).
Whole-body lipolysis is increased in Vpr-Tg and sVpr-treated mice
Accelerated lipolysis is a cardinal metabolic defect in HIV patients (17-19). Lipid kinetic studies using steady-state infusions of isotopes of glycerol and palmitate (17) revealed that fasting total and net lipolysis were increased in Vpr-Tg mice of one line (Fig. 2A) (but not of another line; table S2) and in sVpr-treated mice (Fig. 2B). These Vpr-Tg and sVpr-treated mice had higher fasting respiratory exchange ratios (RERs) than their respective controls under the conditions of the infusion studies, indicating blunted fat oxidation (Fig. 2, C and D) for 4 hours early in the fasting period. However, longer calorimetry under conditions of chronic feeding with regular (n = 11 wild type; n = 10 Vpr-Tg) or high-fat (n = 11 wild type; n = 12 Vpr-Tg) diet, with the mice permitted to move freely, showed no group differences in fat oxidation averaged over 24 hours (table S3).
Fat mass is diminished in Vpr-Tg and sVpr-treated mice
Although food intake was similar in Vpr-Tg and sVpr-treated mice compared to their respective controls (table S4), white adipose tissue (WAT) depots [inguinal fat (IF), perigonadal fat (PGF), and retroperitoneal fat (RPF)] were diminished in Vpr-Tg (Fig. 2E) and sVpr-treated mice (Fig. 2F). There were no group differences in brown adipose tissue mass.
Expression of PPARγ target genes is blunted, whereas that of GR-regulated lipolytic genes is increased, in adipose tissues of Vpr-Tg and sVpr-treated mice
Accelerated lipolysis and diminished fat mass could result from PPARγ co-repression (14) or GR coactivation (12, 13) in adipose tissues. We quantified mRNA expression of PPARγ- and GR-regulated genes critical for fasting adipose metabolism. mRNA levels of the PPARγ targets Pparγ (Fig. 3A), AdipoQ, and Ap2 (Fig. 3B) were diminished in IF of Vpr-Tg. Rosiglitazone did not affect the expression of Pparγ (Fig. 3A), AdipoQ, or Ap2 (fig. S2) in either wild-type or Vpr-Tg IF. mRNA levels of PPARγ-regulated Cap, Glut4, Cd36, Lpl, and Plin1 were also decreased in Vpr-Tg IF (Fig. 3B). Expression of Atgl, a GR-regulated gene responsible for triglyceride lipolysis, was increased, whereas that of Hsl, another GR-regulated gene that regulates diglyceride lipolysis, was decreased, in Vpr-Tg IF (Fig. 3C).
Similar results were noted in PGF, with some differences. mRNA levels of Pparγ (Fig. 3D), AdipoQ, and Ap2 (Fig. 3E) were diminished in Vpr-Tg mice; rosiglitazone modestly increased Pparγ mRNA expression in wild-type but not in Vpr-Tg mice. Vpr's effect on the other PPARγ targets in PGF was more selective: mRNA levels of Cap were decreased, but those of Glut4, Cd36, Lpl, and Plin1 were not different from wild type (Fig. 3E). mRNA expression of both Atgl and Hsl was increased in Vpr-Tg PGF (Fig. 3F).
To demonstrate that circulating Vpr can lead to adipose gene expression changes similar to those caused by Vpr expressed within adipose cells, we compared PPARγ and GR target gene mRNA levels in PGF of sVpr-treated and vehicle (water)-treated mice. Treatment with sVpr for 2 weeks decreased the mRNA levels of the PPARγ targets (Fig. 3G) and increased the mRNA levels of the GR-regulated lipolytic genes (Fig. 3H), similar to the changes observed in Vpr-Tg PGF.
Plasma adiponectin and aP2 protein are diminished, and adipose levels of activated forms of lipolytic enzymes are increased, in Vpr-Tg
Plasma total and high-molecular weight (HMW) adiponectin protein were diminished in Vpr-Tg mice (Fig. 4, A and B). Rosiglitazone increased total and HMW adiponectin protein in both wild-type and Vpr-Tg mice. Plasma aP2 protein was also diminished in Vpr-Tg mice, with no effect of rosiglitazone (Fig. 4C).
Within IF, protein expression of adipose triglyceride lipase (ATGL) showed a trend (P = 0.06) to be increased in Vpr-Tg (Fig. 4D), congruent with increased Atgl mRNA. However, the ratio of phospho-hormone-sensitive lipase (HSL) (Ser563) (indicating a ß-adrenergic-regulated modification that activates catalytic activity) to total HSL was decreased in Vpr-Tg (Fig. 4D). Within PGF, ATGL protein was markedly increased in Vpr-Tg mice (Fig. 4E); furthermore, the ratio of phospho-HSL (Ser563) to total HSL was increased in Vpr-Tg mice, whereas that of phospho-HSL (Ser565) [indicating an adenosine monophosphate-activated protein kinase (AMPK)-regulated modification that does not activate lipolysis] to total HSL was not different (Fig. 4E). In sVpr-treated mice, as in Vpr-Tg mice, ATGL expression was elevated in both IF and PGF, whereas the ratio of phospho-HSL (Ser563) to total HSL was decreased in IF but increased in PGF (Fig. 4F).
Macrophage infiltration is increased and adipocyte morphology is altered in PGF of Vpr-Tg and sVpr-treated mice
Macrophage number was increased (Fig. 4, G and I) and adipocytes were larger (Fig. 4J) in PGF of Vpr-Tg mice. Macrophages, CLSs, and adipocyte size were also increased in PGF of sVpr-treated mice (Fig. 4, G, K, L, and M). Perilipin staining was diminished adjacent to CLSs, indicating increased adipocyte cell death in sVpr-treated mice (Fig. 4H).
Vpr expressed in 3T3-L1 preadipocytes inhibits differentiation via cell cycle block, whereas Vpr expressed in mature adipocytes induces hyperlipolysis
To assess differential effects of Vpr on preadipocytes compared to mature adipocytes (both of which are present in adipose depots), we used a doxycycline-regulated lentiviral expression system in 3T3-L1 cells, inducing either early (during preadipocyte proliferation) or late (after adipocyte differentiation) Vpr expression.
Vpr expression in proliferating preadipocytes completely blocked differentiation (Fig. 5A), associated with decreased expression of genes of the adipocyte development transcriptional cascade, from Pref1/Dlk1 (early proliferation gene) through Pparγ2 (early differentiation gene) to Glut4 (late differentiation gene) (Fig. 5B). This early differentiation block (proximal to Pparγ2 expression) in preadipocytes suggested a Vpr effect distinct from PPARγ co-repression. Flow cytometry revealed that Vpr expression induced cell cycle block at G2-M; cell cycle arrest was attenuated, and preadipocyte differentiation gene expression and lipid accumulation were partially restored with expression of Vpr R80A, a mutant that blunts Vpr's cell cycle arrest function (Fig. 5C and fig. S3). Associated with the G2-M block was a dysregulated increase in cyclin D1 expression (which normally occurs at the G1-S transition) with some increase in cyclin B1 (Fig. 5D) and a sustained elevation of Ccnd1 mRNA levels (encoding cyclin D1) (Fig. 5E) in the Vpr-expressing cells.
Vpr expression induced 48 hours after addition of differentiation medium did not block differentiation (fig. S4); however, ß-adrenergic-stimulated lipolysis was accelerated, as revealed by increased free fatty acid (FFA) release (Fig. 6, A and B) with a similar trend for glycerol release (Fig. 6, A and C).
These results indicated that whereas cell cycle arrest is a pathogenic mechanism in preadipocytes, the effects of Vpr in mature adipocytes involve different mechanisms, possibly PPARγ co-repression or GR coactivation. To test this, we performed chromatin immunoprecipitation (ChIP) assays in 3T3-L1 cells with lentivirus expression induced 72 hours after initiation of adipocyte differentiation, and quantified Vpr's effect on PPARγ-regulated (AdipoQ and Ap2) or GR-regulated (Hsl) gene expression. The levels of AdipoQ and Ap2 promoter DNA immunoprecipitated by anti-PPARγ antibody after rosiglitazone treatment were lower in the Vpr compared to the control (rtTA) condition; in contrast, the level of Hsl promoter DNA immunoprecipitated by anti-GR antibody after dexamethasone treatment was higher in the Vpr condition (Fig. 6D).
Expression of PPARα-regulated genes is down-regulated in liver of both Vpr mouse models
Because fat oxidation is blunted in HIV patients (17) and Vpr-exposed mice during the early fasting period, we quantified the mRNA expression of PPARα-regulated fat oxidation genes in liver of Vpr-Tg and wild-type littermates. mRNA levels of Pparα (Fig. 7A), Cpt1α, Aox, and Lcad (Fig. 7B) were decreased in Vpr-Tg. In liver of sVpr-treated mice, there was a decrease in Lcad mRNA and nonsignificant decreases in mRNA levels of the other oxidation genes (Fig. 7, C and D).
Hepatosteatosis develops in both Vpr mouse models
HIV infection is associated with a high prevalence of hepatosteatosis (20). Hepatic fat was increased in Vpr-Tg (Fig. 7, F and G), with a 1.5-fold elevation of liver triglyceride content (Fig. 7E) and a modest increase in liver weight (Fig. 7H). Fat content and liver weight were also increased in sVpr-treated mice after exposure to sVpr for only 2 weeks (Fig. 7, F, I, and J).
Additional pathogenic pathways contribute to Vpr-induced hepatosteatosis
The rapid development of hepatosteatosis suggested pathogenic pathways in addition to increased FFA flux with impaired hepatic fat oxidation. Decreased plasma adiponectin levels in Vpr-Tg and sVpr-treated mice prompted investigation of its hepatic target AMPK (21), because decreased AMPK activity [resulting in lowered expression of PPARγ coactivator 1α (PGC1α) and thereby of PPARα (22, 23)] can cause hepatosteatosis (24). Activated (Thr172-phosphorylated) AMPK relative to total AMPK was decreased in Vpr-Tg liver (Fig. 7K), as was Pgc1α mRNA (Fig. 7L). Pepck mRNA, whose expression in fasting liver is normally up-regulated by PGC1α (25), was also decreased in Vpr-Tg (Fig. 7L), explaining the low level of PEPCK promoter-driven Vpr transgene expression in liver compared to fat (Fig. 1B).
Intrahepatic apolipoprotein B-triglyceride packaging and transport are regulated by adipose differentiation-related protein (ADRP) [for very low density lipoprotein (VLDL)-triglyceride assembly and storage] and microsomal triglyceride transfer protein (MTP) (for VLDL-triglyceride export). PPARα positively regulates MTP transcription (26), and Mtp knockout induces fatty liver by inhibiting VLDL-triglyceride export (27). Mtp mRNA was diminished in Vpr-Tg liver (Fig. 7L).
Vpr-Tg manifest hepatic insulin resistance and hyperglycemia
Hepatic insulin sensitivity, reflected by insulin-stimulated phospho-Akt (Ser473), was diminished in Vpr-Tg (fig. S5A), associated with elevated postchallenge plasma glucose levels in young and older Vpr-Tg (fig. S5, B and C). Plasma insulin levels were not different (fig. S5D). Fasting plasma triglyceride levels were elevated in Vpr-Tg (fig. S5E), but FFA levels were not different (fig. S5F).
DISCUSSION
Vpr circulates in the blood of HIV patients even after viral-suppressive treatment with ART, and its presence in the circulation of mice is sufficient to recapitulate the characteristic features of adipose and hepatic metabolic defects observed in HIV patients: accelerated lipolysis (17-19), diminished fat mass (7, 28), hepatosteatosis (20), insulin resistance, and hyperglycemia (29). Inhibition of PPAR-regulated signaling in fat and liver is a key mechanism in vivo because Vpr (i) represses PPARγ-regulated genes responsible for adipocyte differentiation, fatty acid transport, and insulin sensitization; (ii) represses PPARα-regulated genes of hepatic fat oxidation and VLDL-triglyceride export; and (iii) lowers adiponectin, leading to decreased liver AMPK-Thr172 phosphorylation and diminished expression of Pgc1α. Concurrently, Vpr promotes the metabolic phenotype by coactivating GR-regulated genes in adipocytes (driving lipolysis) and via cell cycle arrest in preadipocytes (diminishing adipose mass).
Together with in vitro evidence for cellular- and nuclear-transducing activities of virion-free Vpr (16), these data demonstrate that Vpr drives mechanisms that may underlie the metabolic defects in HIV patients. Vpr, produced by infected immune cells within reservoirs in HIV patients on ART (15), can transduce adipose tissues and liver to produce these metabolic defects (Fig. 8). Although the mouse models do not define the cells of origin of Vpr in natural HIV infection or latency, they support a pathogenic process whereby Vpr arriving at adipocytes or hepatocytes alters transcriptional regulation and function in those cells, and that Vpr can be transferred bidirectionally between these cells and the extracellular compartment in vivo. Furthermore, Vpr produced by HIV replicating in CD4+ T cells can act in a paracrine manner on primary human subcutaneous adipocytes, altering mRNA expression of the PPARγ target AdipoQ and of the GR lipolysis-regulating targets Atgl and Hsl in a manner similar to that in IF of Vpr mice (fig. S6). Adverse effects of ART (4) could exacerbate lipodystrophic manifestations and contribute to heterogeneity in the clinical phenotype.
Gene expression studies of fat biopsies from HIV patients corroborate these results. Independent of ART, mRNA levels of PPARγ, adiponectin, and mitochondrial oxidative enzyme genes are reduced in subcutaneous adipose tissue of HIV patients (9). PPARγ positively regulates these genes, indicating that HIV-1 infection per se induces adipose defects via PPARγ repression, consistent with a Vpr effect. Similar gene expression defects occur in fat tissues of HIV-infected, untreated long-term nonprogressors (10) and in mice expressing a 7.7-kb HIV-1 construct (30).
Consistent with varying expression of adipokine genes in different human adipose depots (31) and regional differences in glucocorticoid sensitivity (32), repression of PPARγ-regulated genes was less, whereas activation of GR-regulated lipolytic genes (both Atgl and Hsl) was greater in PGF (analogous to human visceral fat) than in IF (analogous to human subcutaneous fat). In obese humans, lipolysis is higher in visceral than in subcutaneous fat (33), and the flux of fatty acids from this depot directly into the liver could drive hepatosteatosis.
GR signaling, which profoundly enhances ATGL expression (34), would increase ATGL activity and accelerate lipolysis in Vpr-Tg PGF. Protein kinase A activates HSL via Ser563 phosphorylation, which was elevated in PGF but reduced in IF of Vpr-Tg mice. Glucocorticoids also up-regulate HSL expression (35). In mice, expression of ATGL [responsible for basal lipolysis (36)] is increased by fasting, whereas that of HSL is not. The coordinate increase in both ATGL protein and phospho-HSL (Ser563) in PGF of Vpr mice is explained by enhanced glucocorticoid sensitization (12).
Adipose mass is regulated by turnover of preadipocytes through proliferation, differentiation, and apoptosis (37). Vpr expression in 3T3-L1 preadipocytes blocked differentiation, with down-regulation of genes of the relevant transcription factors. Cell cycle arrest at G2-M, associated with continuously elevated expression of cyclin D1 [which interferes with PPARγ-mediated adipogenesis (38)], was a key mechanism of the differentiation block. Expression of the differentiation genes was not fully normalized by Vpr-R80A; hence, it is possible that mitochondria-dependent apoptosis by Vpr (39), which is also mitigated by the R80A mutation (40), contributes to the differentiation block. Expression of Vpr in differentiated adipocytes increased lipolysis and reciprocally altered regulation of PPARγ and GR target genes involved in insulin sensitivity and lipolysis. Diminished WAT mass also occurred without increase in oxidative disposal of fat over 24 hours, indicating that accelerated lipolysis causes ectopic fat deposition in nonadipose organs, as is well described in HIV patients (41-43). Thus, multiple Vpr mechanisms produce lipoatrophy and hyperlipolysis.
Vpr increased macrophage number and CLSs in adipose tissue after exposure to circulating Vpr for only 2 weeks. Adipose tissue macrophages are derived from circulating monocytes; because FFAs are potent monocyte chemoattractants (44), Vpr-induced hyperlipolysis could directly promote macrophage accumulation in adipose depots.
Hepatosteatosis, which is highly prevalent in HIV patients, was induced by Vpr through pathways that likely include impaired fat oxidation (because of down-regulation of hepatic oxidative genes) in the face of increased FFA flux, and impaired clearance of VLDL-triglycerides (because of decreased Mtp expression). A common underlying mechanism is PPARα inhibition both directly by Vpr and indirectly through inhibition of adiponectin expression [leading to blunted hepatic AMPK activity (21) and thereby reduced expression (45) and transcriptional activation (23) of PGC1α (22, 23)]. Down-regulation of AMPK (24) or knockout of Pgc1α or Mtp (27) causes hepatosteatosis in mice.
Fasting hypertriglyceridemia, a hallmark of HIV-associated metabolic defects, was noted in Vpr-Tg mice in the present study [though not in our previous study of a different line of Vpr-Tg mice (46)], suggesting increased VLDL-triglyceride secretion despite diminished Mtp or a defect in VLDL-triglyceride clearance. Both increased secretion and decreased clearance of VLDL-triglycerides occur in HIV patients (18). Fasting plasma FFA levels were not elevated in Vpr-Tg mice, suggesting that increased adipocyte lipolysis is counterbalanced by increased hepatic extraction.
Serum levels of Vpr were not correlated with its quantitative effects on tissue gene or protein expression, lipid kinetics, or fat mass, suggesting a threshold level within the tissues and cells at which Vpr exerts its metabolic effects in tissues. This level cannot be determined from the present studies, but it is at or below that which was associated with the lowest serum Vpr levels in Vpr-Tg, and these are within the range of serum Vpr concentrations in HIV patients. Serum Vpr levels also were not correlated with HIV-1 VL; of note, serum levels of Nef, another HIV protein that is secreted from HIV-infected immune cells and has paracrine effects, are not correlated with HIV viremia (47).
The presence of Vpr in the circulation of optimally treated HIV patients and in adipose tissue and liver of HIV patients at autopsy provides translational relevance to these findings. The current data are limited in demonstrating the critical Vpr mechanisms in mice-full translation would require longitudinal metabolic measurements in blood and tissue specimens of HIV patients on cART with a range of serum Vpr levels and VL. In a cross-sectional analysis, we found no correlation between serum Vpr levels and lipid kinetic rates in 12 hyperlipolytic, hypertriglyceridemic HIV patients on cART with a range of VL, suggesting that Vpr has adipose-restricted effects not reflected quantitatively in serum Vpr concentrations. We infer that Vpr in the serum of patients reflects its production by HIV sequestered in adipose tissue, liver, or other reservoirs but we have not measured Vpr production relative to HIV replication within those tissues or the kinetics of its extracellular transfer. Vpr is likely not the sole pathogenic factor in HIV-associated metabolic disturbances, but it has an independent effect. Confirmation of these mechanisms in patients could pave the way for targeted treatment with small-molecule inhibitors of Vpr, GR antagonists, or dual PPARγ/PPARα agonists.
MATERIALS & METHODS
Study design
The study was designed to test the hypotheses that Vpr affects adipose and liver metabolism in mice and functions as a hormone in mice and HIV patients. Transgenic and pharmacologic mouse models were used to measure lipid kinetics and oxidation, quantitative expression of PPARγ and GR target gene transcripts and their protein products, ChIP, and insulin sensitivity. Hormonal characteristics were determined by measuring serum Vpr levels in mice and stored samples of HIV patients. Sample sizes for all mouse experiments were based on previous experience with Vpr transgenic mice (46). Statistical analyses are described below. Investigators were blinded to all mouse and HIV-infected human sera used for Vpr measurements, and to liver samples used for immunohistochemistry and Oil Red O quantification.
Vpr-Tg mice
Protocols were approved by the Baylor Institutional Animal Care and Use Committee. Vpr-Tg mice expressing PEPCK promoter-driven Vpr under the control of a tetracycline-repressible (tTA) system were constructed at the National Institutes of Health (NIH) (46). Two transgenic lines were rederived at Baylor. Fourteen- to 16-week-old male mice were used in all experiments.
Synthetic Vpr
sVpr was produced by solid-state peptide synthesis, purified, characterized by sequencing and mass spectrometry (MS), and compared to viral Vpr by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblot (48). The stability of the peptide in aqueous solution was confirmed by dynamic light scattering, circular dichroism, and 1H nuclear magnetic resonance spectroscopy (48).
sVpr infusions
Alzet pumps (model 1002, Durect), containing aqueous solution of sVpr or sterile water, were implanted subcutaneously in wild-type mice with delivery rate of 0.25 μl/hour to administer 5 μg of sVpr every 24 hours for 14 days.
Serum Vpr
Serum Vpr was measured in coded samples using ICE (46). This electrokinetic assay uses immobilized monoclonal antibodies (mAbs) to isolate Vpr before separation and online detection by laser-induced fluorescence in samples ≥1 μl. Detection limits were determined using mouse serum standards "spiked" with sVpr. Liquid chromatography-MS and matrix-assisted laser desorption/ionization analysis confirmed that the recovered analyte was comparable to the Vpr used to raise the anti-Vpr antibody for ICE. The specificity of the capture antibody was tested using two-dimensional electrophoresis blots of Vpr, HIV surface and internal antigens, and T cell receptors. The antibody demonstrated reactivity only against Vpr. The sensitivity of the assay was in the range of 1.5 to 2100 pg/ml. Intra- and interassay coefficients of variation were 3.22 and 4.13%, respectively.
Lipid kinetics
Lipid kinetics were measured in Vpr-Tg or wild-type littermates and in wild-type mice receiving sVpr or water (in the latter, 13 days after Alzet placement). Calorimetry (Columbus Instruments) was performed before the kinetic study to measure VO2 and VCO2 under the same conditions as for the isotope infusions (for 4 hours after onset of fasting). The next day, mice received primed (P)-constant IV (I) infusions of [13C1]palmitate (P = 75 μmol/kg, I = 75 μmol/kg per hour) and [2H5]glycerol (P = 140 μmol/kg, I = 140 μmol/kg per hour) for 4 hours. Blood was collected in prechilled Na2EDTA tubes and centrifuged at 4°C, and the plasma was stored at -80°C.
Plasma palmitate isotope ratio was determined on the pentafluorobenzyl
derivative by negative chemical ionization-GC/MS, with selective ion monitoring at mass/charge ratio (m/z) 255 and 256 (Hewlett-Packard); glycerol isotope ratio was measured on the glycerol tripropylester derivative by electron impact-GC/MS, with ion monitoring at m/z 173 to 176. Standard steady-state equations were used to calculate fluxes of palmitate and glycerol; these, with the concentration ratio of plasma palmitate (determined by isotope dilution using [2H2]palmitate) to FFA (Wako), were used to calculate total and net lipolysis (17). Respiratory exchange ratio (RER = VO2/VCO2) provided an index of the fuel substrate oxidized.
Twenty-four-hour calorimetry was also performed in wild-type mice and Vpr-Tg under conditions of chronic feeding with regular chow or high-fat (60%) diet.
v
Effects of Vpr on adipose tissue and liver
Vpr-Tg and wild-type littermates were placed on high-protein diet
(Harlan-Teklad) for 3 weeks to activate the PEPCK/tTA promoter. Subgroups of mice were treated with rosiglitazone (10 mg/kg per day, intraperitoneally) or vehicle for 14 days (dose and duration optimized for adipose expression of Pparγ and Glut4 mRNA). Mice were fasted for 15 hours and euthanized in the morning. Blood and tissues were collected, and plasma was immediately separated. Tissues were snap-frozen for RNA and protein extraction and stored at -80°C or fixed in 10% formalin for immunohistochemistry.
mRNA levels
Total RNA was extracted from liver using TRIzol (Invitrogen) and from adipose tissue using a lipid extraction kit (Qiagen) and transcribed using the RNA-to-cDNA kit (Applied Biosystems), and polymerase chain reaction (PCR) was performed with the TaqMan assay. PPARγ/GR target genes included, in adipose tissues, adiponectin (AdipoQ), adipocyte protein 2 (aP2), Cbl-associated protein (Cap), Glut4, Cd36, perilipin (Plin1), adipocyte triglyceride lipase (Atgl), and hormone-sensitive lipase (Hsl); in liver, carnitine palmitoyl transferase-1α (Cpt1α), acyl coenzyme A (CoA) oxidase (Aox), long-chain acyl CoA dehydrogenase (Lcad), uncoupling protein-2 (Ucp2), and microsomal triglyceride transfer protein (Mtp). Pgk1 mRNA was used for normalization.
Enzyme-linked immunosorbent assay
Plasma concentrations of adiponectin (BioVendor) and aP2 (Invitrogen) were measured by enzyme-linked immunosorbent assay following the manufacturers' protocols.
Immunoblotting
After extraction in radioimmunoprecipitation assay (RIPA) with phosphatase and protease inhibitors, proteins were resolved by SDS-PAGE and identified after transfer by ECL (Thermo Fisher). Primary antibodies for AMPK, phospho-AMPK (Thr172), Akt, phospho-Akt (Ser473), ATGL, HSL, phospho-HSL (Ser563), and phospho-HSL (Ser565) were from Cell Signaling. Immunoblots were scanned, and densitometry was quantified using ImageJ software.
Thin-layer chromatography
Lipids were extracted from 0.2 g of liver, dried under nitrogen, reconstituted in chloroform, and loaded onto a thin-layer chromatography (TLC) plate with standards to identify lipid species. Lipid fractions were visualized using iodine vapor. TLC plates were scanned, and lipid species were quantified by densitometry using ImageJ software.
Oil Red O staining
Cryostat sections of liver were cut, fixed in 10% formalin, and stained with Oil Red O. For each section, three to four pictures of different fields were taken at x20 magnification. Staining was quantified using ImageJ software.
Adipose tissue histomorphometry/immunohistochemistry
Sections were stained for expression of F4/80 and perilipin (Abcam) using standard protocols, and pictures of different fields were taken at x10 to x20 magnification (three mice per group). Adipocytes were counted in two sections per mouse at x10 magnification by observers masked to the treatment. F4/80+ nuclei were counted in nine sections per group. Average cell size was calculated by dividing section area by cell number.
Vpr effects on adipocyte differentiation and function
Vpr complementary DNA (cDNA) was cloned into a tetracycline-inducible lentivirus system (gift of R. Schwartz, University of Houston) with tet-responsive promoter elements (TREs) driving transgene expression and a second lentivirus vector encoding the doxycycline-activated reverse tetracycline transactivator (rtTA). Standard methods were used for virus packaging and propagation. Vector particles were concentrated by ultracentrifugation and titered using Retro-X Kits (Clontech). 3T3-L1 cells and their media for growth, induction, and differentiation were from ZenBio. 3T3-L1 preadipocytes (70% confluence) were infected with lentivirus constructs (TRE-Vpr or TRE and rtTA). For early Vpr expression experiments (Vpr effects in preadipocytes), doxycycline (1 μg/ml) was added 48 hours later (day 0). Differentiation medium (containing insulin, dexamethasone, and PPARγ agonist) was added 48 hours later (day 2) followed by adipocyte maintenance medium. Cells were harvested daily (days 1 to 10), and RNA was isolated (Qiagen). For late Vpr expression experiments (Vpr effects in mature adipocytes), differentiation medium was added on day 2, doxycycline was added to induce transgene expression on day 4, and cells were harvested on day 5 [for quantitative reverse transcription PCR (qRT-PCR) and ChIP] or day 6 (for lipolysis). For qRT-PCR, first-strand cDNA synthesis from total RNA was performed with Superscript III First-Strand Synthesis Kit with oligo(dT) (Invitrogen). qPCR was performed with SYBR Green PCR mix in a Stratagene MX 3000P machine. Levels of Pref1/Glk1, Pparγ2, Ap2, and Glut4 mRNA were measured using validated probes (http://pga.mgh.harvard.edu/primerbank/). Lipolysis was measured on day 6 according to the manufacturer's instructions (ZenBio). Cells were treated in quadruplicate with phosphate-buffered saline (PBS) or CL316243 (ß3-adrenoceptor agonist) for 3 hours. Glycerol and FFA concentrations were measured in the supernatants using a kit (Wako). For ChIP to assay Vpr effect on PPARγ- or GR-regulated adipocyte genes, on day 5, cells were treated with DMSO (vehicle), 1 μM rosiglitazone, or 10 nM dexamethasone for 24 hours and formaldehyde, and ChIP was performed according to the Abcam protocol (http://www.abcam.com/ps/pdf/protocols/x_CHip_protocol.pdf). Immunoprecipitation antibodies were directed against PPARγ (Abcam) or GR (Cell Signaling), with rabbit immunoglobulin G (Abcam) as control. Promoter-targeting primer sequences for qPCR are reported in table S5. For negative controls, primers targeting off-target sites ~2 kb upstream of the respective target peroxisome proliferator gamma response element (PPRE) or glucocorticoid response element (GRE) were used.
Flow cytometry of cell cycle and cyclin expression
3T3-L1 cells were stained with propidium iodide (PI) and cyclin mAbs. Cells (5 x 105) were trypsinized, washed with PBS, fixed with 4 ml of 100% methanol at 4°C overnight, permeabilized in 1 ml of PBS/0.25% Triton on ice for 5 min, washed with PBS/2% fetal bovine serum (FBS), and incubated with mAbs or isotype controls (1 μg/ml) for 1 hour at 4°C. Cells were washed with PBS/2% FBS and incubated with PI (50 μg/ml) (Sigma) and ribonuclease (100 μg/ml) (Sigma) for 1 hour at 4°C. Data were acquired on a Gallios flow cytometer and analyzed with Kaluza 1.2 software (Beckman-Coulter).
Plasma triglyceride levels were measured using the GPO Trinder reagent (Thermo Scientific).
Human subjects
Serum samples for Vpr measurement were collected from HIV-negative subjects and HIV patients on cART who gave informed consent under clinical research protocols approved by the Baylor and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Institutional Review Boards. Sera from ART-naïve and NRTI-treated HIV patients were obtained from deidentified stored samples of participants previously enrolled in an Institutional Review Board-approved vaccine trial.
Statistics
One-way analysis of variance (ANOVA) with Bonferroni's multiple comparison test was used for analysis of serum Vpr levels in HIV-infected patients. All animal experiments involved two-way comparisons between wild-type and experimental conditions; hence, two-tailed, unpaired t tests for unequal variance were used. For lentiviral studies involving comparisons between uninfected control, rtTA control, and the other experimental conditions, or wild-type Vpr compared to Vpr-R80A at different time points, two-way ANOVA with Bonferroni's multiple comparison test was applied. Data are presented as means ± SE. P < 0.05 was considered significant.
|
|
| |
| |
|
|
|