 |
|
|
| |
Evaluation of Transporter and Cytochrome P450-Mediated Drug-Drug Interactions Between Pan-Genotypic HCV NS5A Inhibitor GS-5816 and Phenotypic Probe Drugs
|
| |
| |
(slides below)
15th Intl Wrkshp Clinical Pharm HIV Therapy Drug Interactions Between Direct-Acting anti-HCV Antivirals Sofosbuvir and Ledipasvir and HIV Antiretrovirals - (05/22/14)
Clinically Relevant Interactions Between GS-5816 HCV NS5A Inhibitor and Probe Drugs
15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy, May 19- 21, 2014, Washington, DC
Mark Mascolini
GS-5816, a second-generation HCV NS5A replication complex inhibitor, had potentially clinically relevant interactions with inducers and inhibitors of certain transporters and metabolizing enzymes, according to results of a 5-cohort study of healthy volunteers [1].
Now in phase 2 trials, GS-5816 has in vitro activity against HCV genotypes 1 through 6 and against HCV resistant to first-generation NS5A inhibitors [2]. Gilead Sciences investigators conducted these phase 1 studies to assess potential interactions between GS-5816 and probe substrates (pravastatin, rosuvastatin, or digoxin) or inhibitors and inducers of enzymes and drug transporters (ketoconazole, rifampin, or cyclosporine). Previous work showed that GS-5816 is an inhibitor of transporters OATP, P-gp, and BCRP, and a substrate for P-gp and CYP3A4 and CYP2C8 enzymes.
This open-label single- and multiple-dose crossover study involved 5 cohorts of 76 total healthy volunteers. In each cohort volunteers were randomized to 100 mg of GS-5816 plus the probe agent or to GS-5816 or the probe alone. The researchers calculated geometric least squares means ratios and 90% confidence intervals for area under the concentration-time curve (AUC) and maximum concentration (Cmax) of GS-5816 and probe drugs.
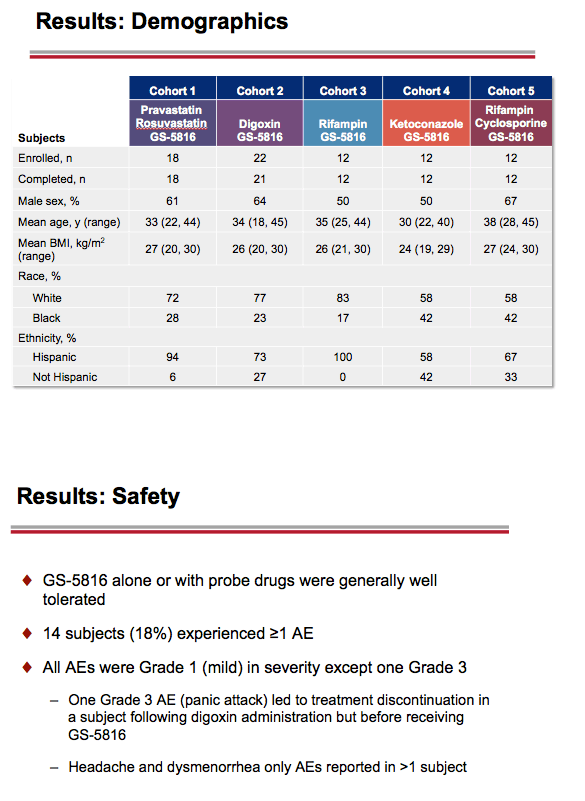
About 60% of volunteers were men, and age averaged about 35 years. Across the five study groups, 58% to 83% were white and 17% to 42% black. Ethnically, most participants in every study group were Hispanic.
Most treatment-emergent adverse events were grade 1. One volunteer had a grade 3 panic attack 2 days after taking a single 0.25-mg dose of digoxin and dropped out of the study.
The following interactions emerged:
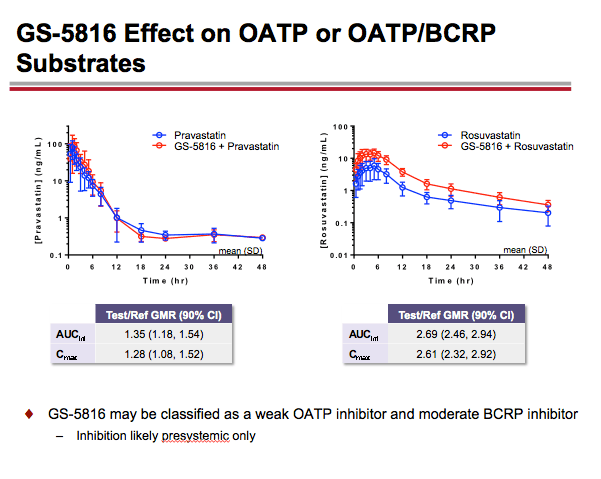
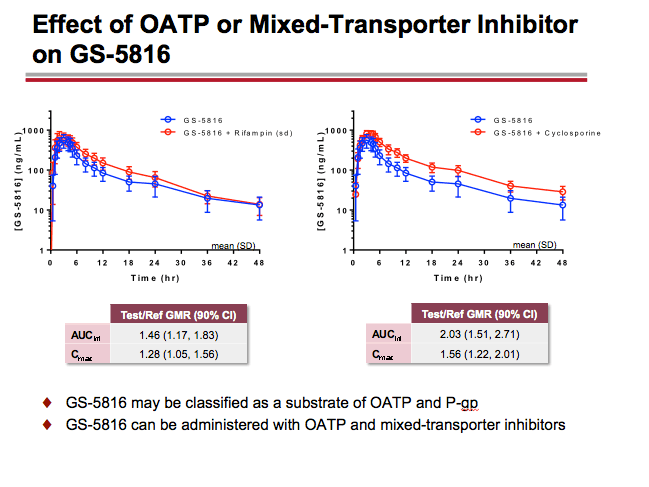
-- GS-5816 taken with the OATP1B1/1B3 substrate pravastatin resulted in approximately a 35% increase in pravastatin AUC and a 28% increase in pravastatin Cmax. GS-5816 taken with the OATP/BCRP substrate rosuvastatin resulted in approximately a 2.7-fold increase in rosuvastatin AUC and a 2.6-fold increase in rosuvastatin Cmax. On the basis of these findings, the Gilead researchers concluded that GS-5816 may be classified as a weak OATP inhibitor and a moderate BCRP inhibitor.
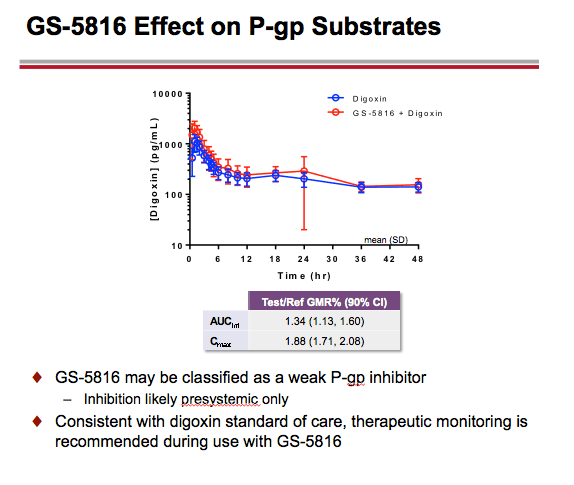
-- GS-5816 taken with the P-gp substrate digoxin resulted in approximately a 34% increase in digoxin AUC and an 88% increase in digoxin Cmax. These findings indicate that therapeutic monitoring would be called for when combining digoxin with GS-5816.
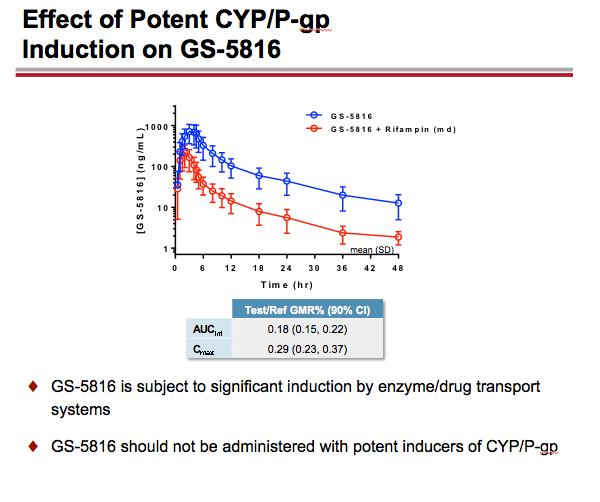
-- GS-5816 taken with the potent CYP3A/CYP2C8/P-gp inducer rifampin resulted in approximately an 82% decrease in GS-5816 AUC and a 71% decrease in GS-5816 Cmax. As a result, GS-5816 should not be used with potent CYP/P-gp inducers.
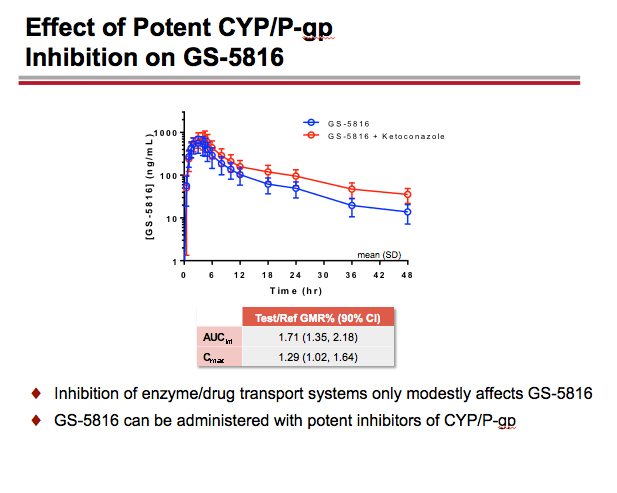
-- GS-5816 taken with the potent CYP3A/CYP2C8/P-gp inhibitor ketoconazole resulted in approximately a 70% increase in GS-5816 AUC and a 29% increase in GS-5816 Cmax. These findings suggest inhibition of enzyme/drug transport systems affects GS-5816 only modestly, so the HCV drug can be taken with potent CYP/Pg-P inhibitors without dose adjustment.
-- GS-5816 taken with the mixed OATP/P-gp/MRP2/CYP3A inhibitor cyclosporine resulted in approximately a doubling in GS-5816 AUC and a 56% increase in GS-5816 Cmax. The Gilead investigators proposed that GS-5816 can be administered with OATP and mixed-transporter inhibitors.
References
1. Mogalian E, German P, Yang C, et al. Evaluation of transporter and cytochrome P450-mediated drug-drug interactions between pan-genotypic HCV NS5A inhibitor GS-5816 and phenotypic probe drugs. 15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy. May 19-21, 2014. Washington, DC. Abstract O_07.
2. Cheng G, Tian Y, Yu M, et al. GS-5816, a second-generation HCV NS5A inhibitor with potent antiviral activity, broad genotypic coverage, and a high resistance barrier. EASL 48th Annual Meeting. April 24-28, 2013. Amsterdam. http://www.natap.org/2013/EASL/EASL_34.htm
-------------------
Evaluation of Transporter and Cytochrome P450-Mediated Drug-Drug Interactions Between Pan-Genotypic HCV NS5A Inhibitor GS-5816 and Phenotypic Probe Drugs
Reported by Jules Levin
15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy, May 19-21, 2014, Washington, DC
Erik Mogalian, Polina German, Chris Yang, Lisa Moorehead, Diana Brainard, John McNally, Jennifer Cuvin, Anita Mathias
Gilead Sciences, Foster City, CA

Program abstract:
Introduction: GS-5816 is an HCV NS5A inhibitor with potent in vitro activity against HCV genotypes 1-6 that is in Phase 2 clinical development for the treatment of chronic HCV infection. In vitro studies demonstrate that GS-5816 is an inhibitor of transporters OATP, P-gp, and BCRP, and a substrate for P-gp and CYP3A4 (minor) and CYP2C8 (minor) enzymes.
This Phase 1 study evaluated the potential of GS-5816 to be a perpetrator or victim of drug-drug interactions using phenotypic probe substrates (pravastatin, rosuvastatin, or digoxin) or inhibitors and inducers of enzymes and drug transporters (ketoconazole, rifampin, or cyclosporine) implicated in its disposition.
Materials & Methods: This was an open-label, single-and multiple-dose, five-cohort crossover study in healthy subjects. Within each cohort, subjects were randomized to receive both test (GS-5816 100 mg + probe) and reference (GS-5816 or probe) treatments. Treatments were separated by appropriate washouts for the respective compounds used in each cohort. Serial blood samples were collected over 72 hours (Cohort 1: pravastatin) or 96 hours (Cohorts 2-5: rosuvastatin, digoxin, cyclosporine, GS-5816) for the PK analyses. Geometric-least squares means ratios and 90% confidence intervals were estimated for AUC and Cmax of probe drugs and GS-5816, as appropriate, and compared against pre-specified lack of PK alteration boundaries of 70 to 143%.
Results: The study enrolled 76 subjects into 5 cohorts. All subjects except one, described below, completed the study. Study drugs were generally safe and well tolerated. Treatment emergent AEs were generally of mild severity (Grade 1). In Cohort 2, a Grade 3 AE (panic attack) was reported 2 days after receiving a single oral dose of digoxin 0.25 mg and before GS-5816 was administered, and led to study discontinuation.
GS-5816 as Perpetrator: Coadministration of GS-5816 with OATP1B1/1B3 substrate pravastatin resulted in AUC and Cmax increases of ~35% and ~28%, respectively. A larger increase in OATP/BCRP substrate rosuvastatin AUC (~170%) and Cmax (~161%) was observed following coadministration with GS-5816. Coadministration of GS-5816 with P-gp substrate digoxin resulted in AUC and Cmax increases of ~34% and ~88%, respectively.
GS-5816 as Victim: Administration of a potent CYP3A/CYP2C8/P-gp inducer rifampin decreased GS-5816 AUC and Cmax by ~82% and ~71%, respectively; whereas, administration of a potent CYP3A/CYP2C8/P-gp inhibitor ketoconazole increased GS-5816 AUC and Cmax by ~70% and ~29%, respectively. Administration of a selective OATP1B1/1B3 inhibitor rifampin increased GS-5816 AUC and Cmax by ~47% and ~28%, respectively. Coadministration of a mixed OATP/P-gp/MRP2/CYP3A inhibitor cyclosporine and GS-5816 did not alter cyclosporine PK but resulted in GS-5816 AUC and Cmax increases of ~102% and ~56%, respectively.
Conclusions: All study treatments were safe and well-tolerated. Clinical results are consistent with preclinical characterization of GS-5816. As a substrate of CYP3A, CYP2C8, and drug transporters such as P-gp and OATP, the disposition of GS-5816 is affected by potent inhibitors and inducers of the enzyme/drug transporter systems. GS-5816 is a weak (P-gp, OATP) to moderate (BCRP) transport inhibitor and clinically relevant drug-drug interactions between GS-5816 and OATP, P-gp, and CYP450 substrates are not anticipated.




|
| |
|
|
|
|
|