| |
Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial
|
| |
| |
Download the PDF here
Download the PDF here
Within cohort 2, PR-null patients with cirrhosis achieved SVR12 rates of 92% (95% CI 74-99, 23/25) with 12 weeks of treatment and 100% (95% CI 85-100, 23/23) with 18 weeks, respectively (appendix); SVR12 rates were similar to those of previously untreated patients with cirrhosis (figure 3). The SVR12 rates for PR-null patients with cirrhosis with HCV genotype 1a or genotype 1b infection were 94% (95% CI 80-99, 31/33) or 100% (95% CI 78-100, 15/15), respectively. Among patients with baseline platelets less than 90 000/μL, 18 of 25 achieved SVR12. Among the five patients with baseline platelets less than 90 000 per μL and baseline albumin less than 3·5 g/dL, four achieved SVR12.
Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial
The Lancet March 21 2015
Eric Lawitz, Edward Gane, Brian Pearlman, Edward Tam, Wayne Ghesquiere, Dominique Guyader, Laurent Alric, Jean-Pierre Bronowicki,
Laura Lester, William Sievert, Reem Ghalib, Luis Balart, Fredrik Sund, Martin Lagging, Frank Dutko, Melissa Shaughnessy, Peggy Hwang,
Anita Y M Howe, Janice Wahl, Michael Robertson, Eliav Barr, Barbara Haber
Summary
Background
There is a high medical need for an interferon-free, all-oral, short-duration therapy for hepatitis C virus (HCV) that is highly effective across diverse patient populations, including patients with cirrhosis or previous null response to pegylated interferon (peginterferon) plus ribavirin (PR-null responders). We aimed to assess the efficacy, safety, and effective treatment duration of grazoprevir (an HCV NS3/4A protease inhibitor) combined with elbasvir (an HCV NS5A inhibitor) with or without ribavirin in patients with HCV genotype 1 infection with baseline characteristics of poor response.
Methods
The C-WORTHY trial is a randomised, open-label phase 2 trial of grazoprevir plus elbasvir with or without ribavirin; here we report findings for two cohorts of previously untreated patients with cirrhosis (cohort 1) and those with previous PR-null response with or without cirrhosis (cohort 2) enrolled in part B of the study. Eligible patients were adults aged 18 years or older with chronic HCV genotype 1 infection and HCV RNA concentrations of 10 000 IU/mL or higher in peripheral blood. We randomly assigned patients to receive grazoprevir (100 mg daily) and elbasvir (50 mg daily) with or without ribavirin for 12 or 18 weeks. Randomisation was done centrally with an interactive voice response system; patients and study investigators were masked to treatment duration up to week 12 but not to treatment allocation. The primary endpoint was the proportion of patients achieving HCV RNA less than 25 IU/mL at 12 weeks after end of treatment (SVR12), assessed by COBAS TaqMan version 2.0. This study is registered with ClinicalTrials.gov, number NCT01717326.
Findings
We describe findings for 253 patients enrolled in cohort 1 (n=123) or cohort 2 (n=130). In cohort 1, we randomly assigned 60 patients to the 12-week regimen (31 with ribavirin and 29 with no ribavirin) and 63 to the 18-week regimen (32 with ribavirin and 31 with no ribavirin); in cohort 2, we randomly assigned 65 patients to the 12-week regimen (32 with ribavirin and 33 with no ribavirin) and 65 to the 18-week regimen (33 with ribavirin and 32 with no ribavirin. High SVR12 rates were achieved irrespective of the use of ribavirin or extension of the treatment duration from 12 to 18 weeks; SVR12 rates ranged from 90% (95% CI 74-98; 28/31; cohort 1, 12 weeks, ribavirin-containing) to 100% (95% CI 89-100; 33/33; cohort 2, 18 weeks, ribavirin-containing). Among patients treated for 12 weeks with grazoprevir plus elbasvir without ribavirin, 97% (95% CI 82-100, 28/29) of patients in cohort 1 and 91% (76-98, 30/33) of patients in cohort 2 achieved SVR12. Adverse events reported in more than 10% of patients were fatigue (66 patients, 26% [95% CI 21-32]), headache (58 patients, 23% [95% CI 18-29]), and asthenia (35 patients, 14% [95% CI 10-19]).
Interpretation
Treatment with grazoprevir plus elbasvir, both with and without ribavirin and for both 12 and 18 weeks' treatment duration, showed high rates of efficacy in previously untreated patients with cirrhosis and previous PR-null responders with and without cirrhosis. These results support the phase 3 development of grazoprevir plus elbasvir.
Funding
Merck & Co, Inc.
Introduction
Globally, 80-185 million people are infected with hepatitis C virus (HCV) according to recent estimates.1, 2, 3 In the coming decades, the number of patients with HCV needing medical care will increase, because of initiatives such as that from the US Centers for Disease Control and Prevention (which has recommended HCV screening for people born between 1945 and 1965) and the universal recognition that many people with HCV have yet to be diagnosed.4, 5, 6 Without therapy, 16% of people with chronic HCV will develop liver cirrhosis within 20 years after infection, and rising to 41% within 30 years.7 In many countries, the number of patients with advanced liver diseases (cirrhosis, decompensated cirrhosis, or hepatocellular carcinoma) is expected to increase as the population infected with HCV ages.8 The number of people infected with HCV who have cirrhosis is projected to peak in the USA at between 626 500 and 1 million in 2015-20.9, 10 Roughly 500 000 deaths in 2010 worldwide were attributable to liver cirrhosis and hepatocellular carcinoma due to HCV.11 Effective therapy and virological cure reduce long-term complications, including hepatic decompensation and hepatocellular carcinoma.12, 13 Together, these facts point to the growing medical need of patients with chronic HCV infection for interventions that will treat and cure their infection.
Available first-line therapies for HCV infection approved for patients with chronic HCV genotype 1 (according to the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver) include sofosbuvir plus pegylated interferon (peginterferon) plus ribavirin, and simeprevir plus peginterferon plus ribavirin, yet these regimens are less effective and worse tolerated in patients with cirrhosis than in previously untreated patients without cirrhosis.14, 15, 16 The sustained virological response rates for sofosbuvir plus peginterferon plus ribavirin in previously untreated patients were 92% for those without cirrhosis and 80% for those with cirrhosis (Metavir fibrosis stage F4).14 The efficacy of simeprevir, peginterferon, and ribavirin was 83-85% in previously untreated patients with HCV genotype 1 and no cirrhosis (Metavir fibrosis stage F0-F2), but only 58-65% in previously untreated patients with cirrhosis (F4) and 53% in patients who had a null response to previous treatment with peginterferon plus ribavirin (PR-null responders).15, 16, 17 The only all-oral regimen currently approved for interferon-ineligible patients with HCV genotype 1 is sofosbuvir plus ribavirin for 24 weeks.18, 19, 20 This regimen, despite a 24-week duration, showed overall efficacy of only 68% (17 of 25 patients) in previously untreated patients without cirrhosis who were chronically infected with HCV genotype 1, but efficacy decreased to 50% (three of six patients) in patients with advanced fibrosis.18 Therapy with peginterferon plus first-line protease inhibitors plus ribavirin in patients with cirrhosis was not well tolerated, with high frequency of serious adverse events.21 Thus, there remains a high medical need for an interferon-free, all-oral, short-duration HCV therapy that is highly effective across all patient populations, including patients with cirrhosis.
Grazoprevir and elbasvir (Merck & Co, Inc, Whitehouse Station, NJ, USA) are highly potent HCV-specific inhibitors of the NS3/4A protease (grazoprevir) and the NS5A protein (elbasvir).22, 23 The combination of grazoprevir and elbasvir has a high barrier to resistance and activity against common resistance-associated variants of HCV.24
In a phase 2 efficacy and safety study, we aimed to assess an all-oral, once-daily combination regimen of grazoprevir plus elbasvir, with or without twice-daily ribavirin, in patients with chronic HCV genotype 1 infection who were previously untreated and who had well compensated cirrhosis, or were previous null responders to peginterferon plus ribavirin with or without cirrhosis. We aimed to examine the need for ribavirin and varying treatment durations, the efficacy and safety of the combination of grazoprevir and elbasvir plus ribavirin, and provide guidance for the design of future studies.
Methods
Study design and participants
C-WORTHY (protocol 035) was a phase 2, parallel-group, multicentre, open-label, international randomised trial enrolling diverse populations; in this report, we describe all patients enrolled in two patient cohorts: cohort 1, consisting of previously untreated patients with well compensated cirrhosis (Child-Pugh A) with HCV genotype 1 infection; and cohort 2, consisting of previously treated patients who were null responders to previous therapy with peginterferon plus ribavirin with or without well compensated cirrhosis (Child-Pugh A) with HCV genotype 1 infection. A companion paper in The Lancet describes efficacy and safety of grazoprevir plus elbasvir with or without ribavirin (treatment duration 8 vs 12 weeks) in two further cohorts of patients (HCV mono-infected and HIV/HCV co-infected patients).25
We enrolled previously untreated adults aged 18 years or older with chronic infection with HCV genotype 1 who had compensated cirrhosis (determined by a liver biopsy, Fibroscan, or Fibrotest plus aspartate aminotransferase to platelet ratio index [APRI]; appendix). Additionally, we enrolled PR-null responder patients (defined as patients who had a less than 2 log10 reduction in HCV RNA at treatment week 12 of a regimen of peginterferon and ribavirin, or who had a less than 1 log10 drop in HCV RNA at treatment week 4) with or without cirrhosis. Inclusion criteria included bodyweight of 50-125 kg, and HCV RNA of at least 10 000 IU/mL in peripheral blood. Exclusion criteria included evidence or history of chronic hepatitis not caused by HCV, positivity for HIV, evidence of hepatocellular carcinoma, decompensated liver disease, previous receipt of any HCV direct-acting antivirals, alanine aminotransferase more than 350 IU/L, aspartate aminotransferase more than 350 IU/L, creatinine clearance less than 50 mL/min, neutrophils less than 1·5 x 103/μL (<1·2 x 103/μL for black people), direct bilirubin more than 1·5 times the upper limit of normal, platelets less than 70 x 103/μL for patients with cirrhosis (<125 x 103/μL for those without cirrhosis), and serum albumin less than 3·0 g/dL for patients with cirrhosis (<3·5 g/dL for those without cirrhosis).
We obtained informed consent from each patient; the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the appropriate institutional review committee.
Randomisation and masking
Randomisation occurred centrally with an interactive voice response system. We randomly assigned patients in a 1:1:1:1 ratio to receive 12 or 18 weeks of treatment with or without ribavirin. All groups were stratified by sub-genotype (1a vs 1 non-a). Additionally, PR-null responder patients were stratified by cirrhosis status (non-cirrhotic vs cirrhotic). The investigators and patients were masked to the duration of therapy from randomisation to week 12, but were not masked to ribavirin allocation; the study funder (including statisticians) was not masked to treatment allocation or duration.
Procedures
We report findings for four treatment groups, to which patients from both cohorts were assigned, within part B of the C-WORTHY study (arms B4-11; figure 1). All groups received grazoprevir (100 mg once daily) plus elbasvir (50 mg once daily) for 12 or 18 weeks with or without ribavirin. Total daily doses of ribavirin were based on the patient's bodyweight (51-65 kg, 800 mg; 66-80 kg, 1000 mg; 81-105 kg, 1200 mg; and >105 kg to 125 kg, 1400 mg) given in divided oral doses in the morning and evening. A central laboratory (Pharmaceutical Product Development, Wilmington, NC, USA) was used to evaluate HCV RNA concentrations.
Plasma HCV RNA concentrations were measured using the Roche COBAS Taqman HCV test, version 2.0, on blood samples drawn from each patient at screening, days 1, 3, 5, 7, weekly between treatment weeks 2 and 12, and after end of treatment at follow-up weeks 2, 4, 8, 12, and 24. The test's lower limit of quantification is 25 IU/mL and its limit of detection is 15·1 IU/mL. We defined virological breakthrough to be confirmed HCV RNA concentration of at least 25 IU/mL after being less than 25 IU/mL previously. We defined relapse as a confirmed HCV RNA concentration of at least 25 IU/mL following end of all study therapy after becoming undetectable (HCV RNA <15·1 IU/mL) at end of treatment. For both virological breakthroughs and relapses, we defined confirmation as a result of HCV RNA concentration of at least 25 IU/mL from a separate blood draw repeated within 2 weeks. Patients did not continue to take study drugs if they had confirmed virological breakthrough or relapse. Patients were followed up for 24 weeks after the end of all study therapy for efficacy outcomes, and 14 days after the end of all study therapy for safety analyses.
Outcomes
The primary efficacy objective of this study was to evaluate the sustained virological response rate 12 weeks after the end of all study therapy (SVR12) for each of the treatment groups. The SVR12 rate was defined as the proportion of patients with HCV RNA less than 25 IU/mL at 12 weeks after the end of all study therapy. We also aimed to assess events of clinical interest recorded through adverse event case report forms including overdose of study drugs, first instance of alanine aminotransferase or aspartate aminotransferase more than 500 IU/L, first instance of alanine aminotransferase or aspartate aminotransferase more than three times baseline and more than 100 IU/L, and first instance of alkaline phosphatase more than three times the upper limit of normal.
Statistical analysis
We did intention-to-treat analyses in all patients who were randomly assigned and received at least one dose of study drug. We calculated two-sided 95% CIs using the Clopper-Pearson method for the SVR12 for each group separately. We did exploratory subgroup analyses, which were not pre-planned, to assess the consistency of response across various demographic and baseline clinical characteristics for patients who received ribavirin and for those who did not receive ribavirin. We calculated two-sided 95% CIs for the difference in the proportion of patients who achieved SVR12 using the Miettinen and Nurminen method.26 There was no formal efficacy hypothesis testing done in this study. For power analysis we used two-sided 95% CIs for SVR12 rates under varying assumptions of the number of successes, assuming a rate of protocol violation of roughly 10%; the results of this analysis suggested a goal of 30 participants from different patient populations to be randomly assigned into each of the various treatment regimens. This study is registered with ClinicalTrials.gov, number NCT01717326.
Role of the funding source
The funder contributed to trial management, data collection, statistical analyses, writing, and internal review of the report. All coauthors had access to the study data, reviewed and approved the final report, and take full responsibility for the veracity of the data and statistical analysis. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Study enrolment began in Feb 27, 2013, and data collection for SVR12 concluded in July 2, 2014. A total of 471 patients were enrolled in the C-WORTHY study; here, we describe findings for 253 patients enrolled in part B of the study who were previously untreated and had cirrhosis (cohort 1, n=123, arms B4-7) or who were PR-null, with or without cirrhosis (cohort 2, n=130, arms B8-11). In cohort 1, we randomly assigned 60 patients to the 12-week regimen (31 with ribavirin [arm B4 of the trial] and 29 with no ribavirin [arm B5]) and 63 to the 18-week regimen (32 with ribavirin [arm B6] and 31 with no ribavirin [arm B7]); in cohort 2, we randomly assigned 65 patients to the 12-week regimen (32 with ribavirin [arm B8] and 33 with no ribavirin [arm B9]) and 65 to the 18-week regimen (33 with ribavirin [arm B10] and 32 with no ribavirin [arm B11]; Figure 1, Figure 2). There were no deviations from the protocol in the conduct of the study as a whole (eg, objectives or hypotheses, or in stratification).
Table 1 shows patient demographics. Most patients in this study had cirrhosis (170 patients, 67%), were white (233 patients, 92%), and were male (148 patients, 58%), with a median age of 56 years. 192 (76%) patients had baseline HCV RNA of more than 2 000 000 IU/mL, 163 (64%) had HCV genotype G1a, and 206 (81%) were IFNL3 non-CC (CT or TT genotypes at the single nucleotide polymorphism rs12979860; appendix).
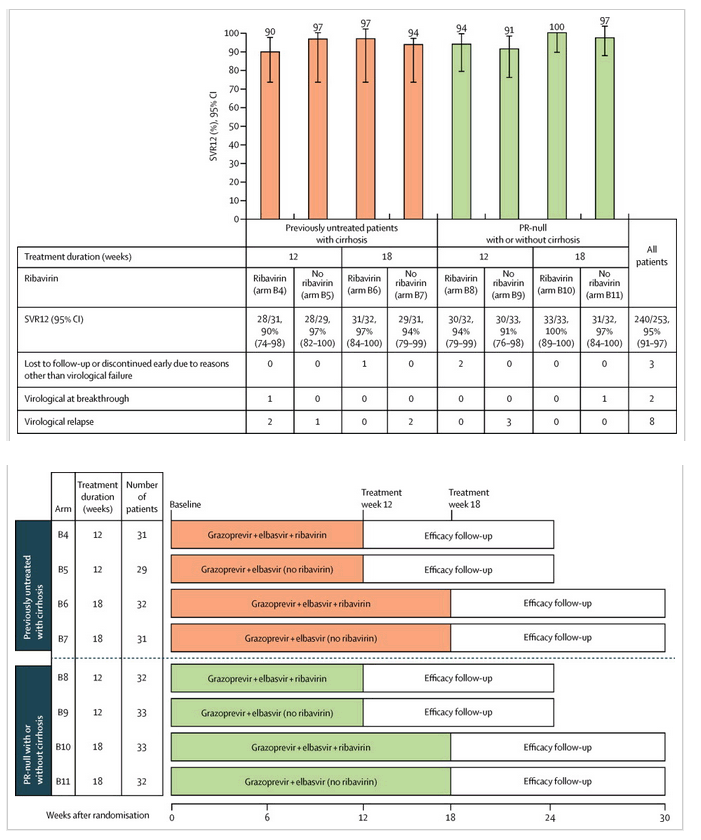
In cohort 1, SVR12 rates ranged from 90% (95% CI 74-98 [28/31] in arm B4) to 97% (95% CI 82-100 [28/29] in arm B5 and 95% CI 84-100 [31/32] in arm B6; figure 3). One patient (arm B6) discontinued treatment early because of adverse events, one patient (arm B4) had virological breakthrough, and five patients (arms 4, 5, and 7) had virological relapse. In cohort 2, SVR12 rates ranged from 91% (95% CI 76-98 [30/33] in arm B9) to 100% (95% CI 89-100 [33/33] in arm B10; figure 3). One patient (arm B8) discontinued because of adverse advents and one patient died (arm B8), one patient (arm B11) had virological breakthrough, and three patients (arm B9) had virological relapse. The overall rate of virological failure (either breakthrough or relapse) was 4% (ten of 253 patients).
Figure 3
Rates of sustained virological response after 12 weeks of follow-up
We defined sustained virological response after 12 weeks of follow-up defined as hepatitis C virus RNA concentration less than 25 IU/mL 12 weeks after the end of all study therapy in the intention-to-treat population (all randomly assigned patients who received at least one dose of study treatment). Blood samples were drawn from patients at screening, days 1, 3, 5, 7, weekly between treatment weeks 2 and 12, and after end of treatment at follow-up weeks 2, 4, 8, 12, and 24.
Sustained virological response rates for all treatment groups in both cohorts exceeded 90%, including in patients treated with the simplest, shortest regimen (arms B5 and B9, 12 weeks, ribavirin-free). Among the previously untreated cirrhotic population, the rate of virological failure was 5% (six of 123 patients) and in the PR-null population, the rate of virological failure was 3% (four of 130).
Within cohort 2, PR-null patients with cirrhosis achieved SVR12 rates of 92% (95% CI 74-99, 23/25) with 12 weeks of treatment and 100% (95% CI 85-100, 23/23) with 18 weeks, respectively (appendix); SVR12 rates were similar to those of previously untreated patients with cirrhosis (figure 3). The SVR12 rates for PR-null patients with cirrhosis with HCV genotype 1a or genotype 1b infection were 94% (95% CI 80-99, 31/33) or 100% (95% CI 78-100, 15/15), respectively. Among patients with baseline platelets less than 90 000/μL, 18 of 25 achieved SVR12. Among the five patients with baseline platelets less than 90 000 per μL and baseline albumin less than 3·5 g/dL, four achieved SVR12.
Subgroup analysis did not show a significant benefit of adding ribavirin to the regimen of grazoprevir plus elbasvir (table 2). Ribavirin did not significantly improve efficacy in patients with cirrhosis, in PR-null responders, or for the factors of treatment duration, age, sex, HCV subgenotype, or body-mass index of 30 kg/m2 or higher. The SVR12 rates were similar in patients with a very high viral load (>10 000 000 IU/mL) with or without ribavirin, suggesting that ribavirin might not have additional benefit in these patients. We noted small numerical differences in some subgroups, possibly resulting from small sample sizes. For example, addition of ribavirin in patients with IFNL3 (also known as IL28B) CC resulted in a lower SVR12 of 88% (15/17) compared with 100% (23/23) without ribavirin, but the 95% CI of the treatment difference crossed 0 (difference -11·8, 95% CI -34·7 to 3·9). A similar small difference was noted for non-white people but the numbers were small and the differences fell within the 95% CIs.
The regimens of grazoprevir plus elbasvir with or without ribavirin were generally well tolerated (table 3, appendix). Common adverse events were fatigue, headache, and asthenia, and were characterised as mild to moderate in intensity. Overall, drug-related adverse events occurred in 159 (63%) of 253 patients. Serious adverse events occurred in seven (3%) of 253 patients, one (<1%; arm B11) of which (abdominal pain) was drug related according to the investigator. Few patients (three [1%] of 253) discontinued treatment because of adverse events (table 3). We noted no grade 4 elevations of ALT and no discontinuations due to ALT elevations. One PR-null responder patient treated for 18 weeks with grazoprevir, elbasvir, and ribavirin (arm B10) had a grade 3 increase in alanine aminotransferase to more than 170 IU/L at 12 weeks of treatment, which resolved without interruption of therapy. The frequency of drug-related adverse events (91 [71%] patients) and discontinuations due to adverse events (three [2%] patients) were higher in the regimens containing ribavirin than in regimens without ribavirin (68 [54%] and no [0%] patients, respectively; appendix). Additionally, the incidence of bilirubin elevation or haemoglobin decrease was higher in patients receiving a regimen containing ribavirin than in those not receiving ribavirin (appendix). No patients had decompensation of liver function while on therapy or during the 12 weeks after end of therapy.
We examined the HCV RNA sequences from patients before treatment and at virological failure for potential resistance-associated variants (RAVs). At baseline, we detected NS3 RAVs in 32% of patients (79 of 248; appendix). These RAVs included those showing more than five-times increased resistance to grazoprevir (eg, D168E) and RAVS known to decrease efficacy of other protease inhibitors (eg, Q80K/R and S122A/G/R; appendix).14, 15 SVR12 was achieved in 92% (73 of 79) of patients with NS3 RAVS, whereas among the 169 patients with wild type at baseline, 96% (163 of 169) achieved SVR12 (appendix). NS5A RAVs were noted at baseline in 14% of patients (34 of 243); the most common variants were at positions 31 and 93 reported in 7% (16/243) and 4% (9/243) of samples, respectively (appendix). Among patients with NS5A RAVS, SVR12 was achieved in 82% (28 of 34), whereas among the 209 patients with wild type NS5A at baseline 97% (203 of 209) achieved SVR12 (appendix). At the time of virological failure, RAVs for NS3 or NS5A were detected in most patients (eight of ten); the most common RAVs detected were NS3:Y56H, A156T/G/V and D168A/Y, and NS5A:M28T, Q30L/R, L31M, and Y93H/N (appendix). Baseline characteristics for the patients who had virological failure are shown in the appendix.
Discussion
In this open-label phase 2 study of an HCV NS3 protease inhibitor (grazoprevir) plus an HCV NS5A inhibitor (elbasvir) with or without ribavirin in HCV genotype 1-infected populations that are difficult to cure with HCV therapy (patients with well compensated cirrhosis and null responder patients with or without well compensated cirrhosis), high rates of efficacy were shown across all groups irrespective of the inclusion of ribavirin or extension of treatment duration from 12 to 18 weeks. Specifically, a regimen of 12 weeks of grazoprevir plus elbasvir without ribavirin showed efficacy of 97% in previously untreated patients with cirrhosis, 91% in null responder patients with or without cirrhosis, and 92% in null responder patients with cirrhosis. The rate of virological failure with grazoprevir plus elbasvir with or without ribavirin was low (4%).
For many regimens, even those that are highly potent in a previously untreated, non-cirrhotic population, efficacy can substantially decrease in patients with cirrhosis and particularly in patients with cirrhosis and previous null response to peginterferon plus ribavirin.15, 16, 27 Other combinations of NS3 protease inhibitor plus NS5A inhibitor have not achieved a high sustained virological response, either because of baseline factors such as cirrhosis or because of differential potency in both genotype 1a and genotype 1b. Even a potent nucleotide regimen such as sofosbuvir plus peginterferon and ribavirin has decreased efficacy in patients with cirrhosis, and especially in genotype 1b-infected patients (panel). An ideal regimen is safe and effective across diverse populations, irrespective of baseline factors such as cirrhosis, previous treatment history, sub-genotype, or other demographic factors. Additionally, the success of HCV therapy in these hard-to-cure populations, particularly null responder patients with cirrhosis, provides a benchmark that can be used to measure the effectiveness of a regimen.
Panel
Research in Context
Systematic review
We searched PubMed and meeting abstracts (European Association for the Study of the Liver and American Association for the Study of Liver Diseases) up to Sept 5, 2014, for clinical trials done in patients infected with hepatitis C virus (HCV) genotype 1, with the terms "HCV" and "hepatitis C". The HCV landscape continues to evolve. Direct-acting antivirals have been approved to treat HCV genotype 1 infection in regimens with pegylated interferon and ribavirin such as simeprevir and sofosbuvir and in all-oral regimens such as sofosbuvir with ribavirin. Nonetheless, some hard-to-treat populations particularly those with cirrhosis and those who have had a null response to previous therapy have lower response rates.
Interpretation
Our study findings suggest that a regimen combining two direct-acting antivirals (grazoprevir and elbasvir) without a nucleotide can achieve sustained virological response of more than 90% with acceptable safety and tolerability profile in patients with cirrhosis or previous null response, as well as patients with genotype 1a or genotype 1b infection. Other dual regimens of an NS3/4A protease inhibitor and an NS5A inhibitor have not shown sustained viral response rates above 90% in some patient populations; these findings might be related to patient baseline factors such as cirrhosis or differential potency across subgenotypes (eg, genotype 1a vs genotype 1b).28 The results of this trial are consistent with the results of a nucleotide-containing 12-week regimen of once-daily sofosbuvir and ledipasvir in which sustained virological response was achieved in 94% of patients who had previous null response to pegylated interferon plus ribavirin. In this study patients had high rates of sustained viral response. However, even in patients who also received a nucleotide polymerase inhibitor (sofosbuvir), we noted a difference in efficacy for patients with genotype 1a infections compared with those with genotype 1b infection (sustained virological response 95% vs 87%) and presence and absence of cirrhosis (sustained virological response 86% vs 95%).28 In summary, this combination of grazoprevir and elbasvir had consistently high sustained virological response with both a 12-week and an 18-week regimen in patients with cirrhosis or previous null response including patients with genotype 1a or genotype 1b infection. This regimen in these difficult-to-cure patient populations showed a low rate of virological failure of just 7% (genotype 1a) or 2% (genotype 1b).
Across subgroups, the 12-week, ribavirin-free regimen resulted in high rates of SVR12. This high efficacy was consistent irrespective of baseline factors such as cirrhosis, previous null response to peginterferon plus ribavirin, HCV sub-genotype, body-mass index of 30 kg/m2 or higher, and high viral load. Patients with genotype 1a or genotype 1b HCV infection achieved SVR12 rates of more than 90% with or without ribavirin. The rates of virological failure among previously untreated patients with well compensated cirrhosis and null responder patients with or without well compensated cirrhosis (excluding discontinuations due to reasons other than virological failure) were eight (5%) of 163 in the genotype 1a population and one (1%) of 87 in the genotype 1b population.
Several investigational all-oral regimens with and without ribavirin have begun to address the deficiencies of recommended first-line therapies, but these new regimens still have limitations. For example, in a trial28 of previously treated patients, sofosbuvir plus ledipasvir (Gilead Sciences, Inc, Foster City, CA, USA) with or without ribavirin showed overall efficacy of 94-99%. Nevertheless, the 12-week ribavirin free regimen of sofosbuvir and ledipasvir resulted in an SVR12 of 86% in patients with cirrhosis and 87% in genotype 1b patients.28 A regimen of ABT-450 plus ritonavir plus ombitasvir plus dasabuvir (AbbVie, North Chicago, IL, USA) plus ribavirin showed efficacy in patients with cirrhosis of 92% or 96% after 12 or 24 weeks of treatment, respectively, yet had increased adverse events.27 A decrease in efficacy was noted with advanced liver disease for the regimen of ABT-450 plus ritonavir, ombitasvir, dasabuvir, and ribavirin for 12 weeks, in which sustained virological response ranged from 82-87% in sample sizes of 25-56 patients with cirrhosis and platelet counts less than 90 x 109/L, albumin less than 35 g/L, α fetoprotein ≥ 20 ng/mL, or Child-Pugh score of 6.27 Similarly, a 12-week regimen of simeprevir plus sofosbuvir with or without ribavirin resulted in SVR12 in 89% of well compensated patients with cirrhosis (excluding patients who did not respond to treatment for reasons other than virological failure).29 The results of this study show that a regimen consisting of a highly potent NS3/4A protease inhibitor (grazoprevir) plus NS5A inhibitor (elbasvir) can achieve consistently high SVR12 rates, even among individuals with factors associated with poor response to other therapies.
The regimen of grazoprevir plus elbasvir with or without ribavirin was generally well tolerated. The incidence of serious adverse events and discontinuations due to adverse events was low (3% and 1%, respectively) with grazoprevir plus elbasvir with or without ribavirin. The incidence of drug-related adverse events and discontinuations due to adverse events were higher in the ribavirin-containing groups than in the groups without ribavirin. The frequency of adverse events leading to discontinuation of study medication was <1% (two of 253) of the total patient population, 2% (two of 128) in the ribavirin-containing groups, and 0% (0 of 125) in the ribavirin-free groups. Furthermore, anaemia and transient elevations in bilirubin were reduced in the ribavirin-free groups. Ribavirin was also a major contributor to adverse events, similar to the findings for sofosbuvir plus ledipasvir.30 These results suggest that ideal regimens should be ribavirin-free.
The presence of NS3 RAVs at baseline did not significantly affect the efficacy of grazoprevir plus elbasvir with or without ribavirin, although we noted some effect of NS5A baseline RAVs on SVR12. Most (92%, 73 of 79) of the patients with NS3 baseline RAVs achieved SVR12. Among the 34 patients with NS5 RAVs at baseline, 82% (28 of 34) achieved SVR12. At the time of virological failure, RAVs at either NS3 or NS5A were reported in eight of ten patients. These results might have implications for salvage therapy of the low percentage of patients who virologically do not respond to grazoprevir plus elbasvir.
This study of grazoprevir plus elbasvir with or without ribavirin had several limitations. First, this study was not statistically powered to determine the precise contribution of ribavirin or extended treatment duration, nor did this study include medically fragile patients that are likely to be treated in clinical practice. Second, the sample sizes in some of the subgroups (eg, non-white and Hispanic or Latino patients) were small. Therefore, with respect to the subgroup analysis, a study with more patients might have shown some significant differences between subgroups. Third, the patients with cirrhosis in this study were well compensated. Thus, the results with grazoprevir and elbasvir might not be applicable to patients with decompensated cirrhosis.
Furthermore, as larger numbers of patients are studied, a multifactorial analysis can be done to provide insight into the effect on efficacy of the convergence of multiple negative factors. Because no comparison of treatment efficacies between groups was done in this trial, the findings should be treated as hypothesis generating to inform the design of future studies.
This study has shaped the phase 3 development programme for grazoprevir and elbasvir. Although a large number of phase 3 studies have been initiated that will treat patients with a two-drug, ribavirin-free regimen of 12 weeks' duration, careful attention clearly needs to be given to the precise contribution of ribavirin and extended treatment duration in addition to inclusion of medically fragile patients in these studies. The phase 3 programme includes studies with the two-drug, 12-week ribavirin-free regimen in previously untreated patients with and without cirrhosis, as well as patients with HIV co-infection, renal failure, previous intravenous drug use, advanced cirrhosis (Childs Pugh B), and inherited blood disorders to assess efficacy and safety across diverse patient populations. A large adequately powered study in previously treated patients with and without cirrhosis will fully assess the need for ribavirin and extended duration (12 and 16 weeks).
In conclusion, our study findings show that a highly potent, all-oral, simple once-daily combination of a protease inhibitor (grazoprevir) plus NS5A inhibitor (elbasvir) could achieve high rates of efficacy across populations that were particularly difficult to cure with HCV therapy. These data support the phase 3 development of the combination regimen of grazoprevir plus elbasvir.
|
|
| |
| |
|
|
|