| |
Ombitasvir/paritaprevir/r and dasabuvir plus ribavirin in HCV genotype 1-infected patients on methadone or buprenorphine
|
| |
| |
Download the PDF here
Journal of Hepatology Aug 2015
Jacob Lalezari1,, J. Greg Sullivan2, Peter Varunok3, Edward Galen4, Kris V. Kowdley5,
Vinod Rustgi6, Humberto Aguilar7, Franco Felizarta8, Barbara McGovern9, Martin King9,
Akshanth R. Polepally9, Daniel E. Cohen9
1Quest Clinical Research, San Francisco, CA, United States; 2Parkway Medical Center, Birmingham, AL, United States; 3Premier Medical Group of
the Hudson Valley, PC, Poughkeepsie, NY, United States; 4Northwest Gastroenterology Clinic, Portland, OR, United States; 5Digestive Disease
Institute, Virginia Mason Medical Center, Seattle, WA, United States; 6Thomas E. Starzl Transplantation Institute, UPMC Montefiore, Pittsburgh,
PA, United States; 7Louisiana Research Center, LLC, Shreveport, LA, United States; 8Private Practice, Bakersfield, CA, United States; 9AbbVie Inc.,
North Chicago, IL, United States
Background & Aims
Hepatitis C virus (HCV)-infected patients with a history of injection drug use have low rates of initiation and completion of interferon-based therapies. This study evaluated efficacy, safety, and pharmacokinetics of a 12-week all-oral regimen of ombitasvir/paritaprevir/ritonavir and dasabuvir + ribavirin in HCV genotype 1-infected patients on stable opioid replacement therapy.
Methods
This was a phase II, multicenter, open-label, single-arm study in treatment-naïve or peginterferon/ribavirin treatment-experienced HCV genotype 1-infected patients on methadone or buprenorphine ± naloxone. Patients received 12 weeks of co-formulated ombitasvir/paritaprevir/ritonavir (25 mg/150 mg/100 mg once daily) and dasabuvir (250 mg twice daily) + weight-based ribavirin. The primary efficacy endpoint was sustained virologic response 12 weeks post-treatment.
Results
Thirty-eight non-cirrhotic patients on chronic methadone (n = 19) or buprenorphine (n = 19) were enrolled. A total of 37 patients (97.4%) had a sustained virologic response 12 weeks post-treatment. No patient had a viral breakthrough or relapse. One patient discontinued due to serious adverse events unrelated to study drug (cerebrovascular accident and sarcoma). The most frequent adverse events were nausea, fatigue, and headache. Eight patients had on-treatment hemoglobin concentrations <10 g/dl. Pharmacokinetic analyses indicated no clinically meaningful impact of methadone or buprenorphine on ombitasvir, paritaprevir, ritonavir, dasabuvir, or dasabuvir M1 metabolite exposures. No dose adjustments of methadone or buprenorphine were required.
Conclusions
The interferon-free regimen of ombitasvir/paritaprevir/ritonavir and dasabuvir + ribavirin for 12 weeks was well tolerated and achieved sustained virologic response in 97.4% of patients on opioid substitution therapy in this study. This all-oral regimen may provide an effective alternative to interferon-based therapies for HCV-infected patients with a history of injection drug use.
Introduction
Injection drug use is the main source of hepatitis C virus (HCV) infections in industrialized nations [1]. Worldwide, approximately 10 million people who inject drugs (PWID) have been infected [2], with a global prevalence among long-term injection drug users of 64-94% [[3], [4]]. Compounding this problem, rates of treatment initiation for HCV-infected PWID have been reported to be under 10% [[5], [6], [7]]. Among the small portion of PWID who do initiate treatment, response rates are limited by a high frequency of discontinuation related to interferon toxicity [[8], [9], [10], [11], [12], [13]]. Thus, interferon-free regimens of direct-acting antivirals with improved efficacy and tolerability may be crucial in increasing the rate of successful treatment in this population.
Paritaprevir (formerly ABT-450) is a NS3/4A protease inhibitor that was identified by AbbVie and Enanta as a lead compound for clinical development. Co-administration with ritonavir, an inhibitor of the cytochrome P-450 enzyme CYP3A4, increases the peak, trough, and overall plasma concentrations of paritaprevir, resulting in high paritaprevir exposures with once daily dosing [14]. When administered with ritonavir to previously untreated genotype 1-infected patients, 3 days of paritaprevir monotherapy decreased mean plasma HCV RNA levels by approximately 4 log10 IU/ml [15]. Ombitasvir (formerly ABT-267) is a NS5A inhibitor; 3 days of ombitasvir monotherapy decreased mean HCV RNA levels by approximately 3 log10 IU/ml [16]. Ombitasvir, paritaprevir, and ritonavir have been co-formulated into a single fixed-dose tablet (ombitasvir/paritaprevir/r). Dasabuvir (formerly ABT-333) is a non-nucleoside NS5B polymerase inhibitor; 3 days of dasabuvir monotherapy decreased mean HCV RNA levels by approximately 1 log10 IU/ml [17].
The combination of these three direct-acting antiviral agents plus ribavirin was well tolerated in clinical trials, and resulted in high sustained virologic response rates in genotype 1-infected patients in the absence of exogenous interferon [[18], [19], [20]]. In phase III trials, 12 weeks of treatment with ombitasvir/paritaprevir/r and dasabuvir plus ribavirin resulted in sustained virologic response rates 12 weeks post-treatment of 96% in both previously untreated and treatment-experienced patients without cirrhosis [[18], [20]]. One percent or less of patients in these trials discontinued treatment due to adverse events [[18], [20]]. The study presented here examined the efficacy, safety, and pharmacokinetics of this multi-targeted regimen in HCV genotype 1-infected patients on chronic opioid replacement therapy with either methadone or buprenorphine.
Patients and methods
Patients
Patients were screened and enrolled at eight sites in the United States beginning in April 2013. Eligible patients were 18-70 years of age and had a body mass index ≥18 and <38 kg/m2, chronic HCV genotype 1 infection, plasma HCV RNA >10,000 IU/ml, and absence of cirrhosis. Patients were treatment-naïve or peginterferon/ribavirin treatment-experienced, and on a stable opioid replacement therapy of methadone or buprenorphine ± naloxone for at least 6 months before screening. Patients had no evidence of HIV or hepatitis B coinfection and no liver disease due to causes other than HCV. All patients signed an informed consent form. The study was conducted in accordance with the protocol, International Conference on Harmonisation guidelines, applicable regulations and guidelines governing clinical study conduct, and the ethical principles that have their origin in the Declaration of Helsinki.
Study design
This was a phase II, multicenter, open-label, single-arm study. All patients received ombitasvir/paritaprevir/r 25 mg/150 mg/100 mg once daily and dasabuvir 250 mg twice daily for 12 weeks. Ribavirin was dosed twice daily at 1000 mg total daily dose for patients <75 kg or 1200 mg total daily dose for patients ≥75 kg. Patients were followed for 48 weeks after the end of the treatment period. The study protocol was developed by the investigators and AbbVie. All authors had access to the data and participated in data analysis and preparation of the manuscript.
Efficacy assessments
Plasma HCV RNA levels were determined by a central laboratory using the Roche COBAS® TaqMan® real-time reverse transcriptase-polymerase chain reaction assay v2.0. The lower limit of detection (LLOD) of this assay is 15 IU/ml and the lower limit of quantitation (LLOQ) is 25 IU/ml. HCV genotype and subtype were assessed from a plasma sample collected at screening using the Versant® HCV Genotype Inno-LiPA Assay, version 2.0 (LiPA; Siemens Healthcare Diagnostics, Tarrytown, NY). Samples for HCV RNA measurement were collected at screening, study day 1, weeks 1, 2, 4, 6, 8, 10, and 12 (or end of treatment), and at post-treatment weeks 2, 4, 8, 12, 24, 36, and 48 (or discontinuation). Treatment was to be stopped if any of the following criteria were met: a confirmed HCV RNA increase from nadir at any time point during treatment, failure to achieve HCV RNA < LLOQ by week 6, or confirmed HCV RNA ≥ LLOQ at any point during treatment after HCV RNA < LLOQ.
Safety analyses
Adverse events were collected from the time of study drug administration until 30 days following the end of study drug administration. Serious adverse events were collected from the time of signing of the informed consent form through the end of the study. Adverse events were coded using the Medical Dictionary for Regulatory Activities (MedDRA®). The investigator assessed the severity of each adverse event and its relationship to the use of study drug. Clinical laboratory testing occurred at all treatment period visits and at post-treatment weeks 4 and 48, or at the time of study discontinuation.
Steady-state pharmacokinetic analyses
Blood samples for the determination of dasabuvir, dasabuvir M1 metabolite, ombitasvir, paritaprevir, ritonavir, and ribavirin concentrations were collected approximately 2, 4, 6, and 24 hours post-dosing at a visit at least 2 weeks after the start of treatment, from those patients who gave consent for intensive pharmacokinetic sampling. The Medication Event Monitoring System (MEMS) caps were used to obtain daily dosing histories for dasabuvir, ombitasvir/paritaprevir/ritonavir, and ribavirin for all patients. Plasma samples were processed and assayed according to validated bioanalytical methods. Plasma concentrations of dasabuvir, dasabuvir M1 metabolite, ombitasvir, paritaprevir, ritonavir, and ribavirin were determined using a validated liquid chromatography method with tandem mass spectrometric detection with LLOQ of 4.58, 4.77, 0.462, 0.601, 4.93, and 98.1 ng/ml, respectively. Individual blood concentrations of S-methadone, R-methadone, buprenorphine, norbuprenorphine, and naloxone were not measured.
Statistical assessments
The primary endpoint was the percentage of patients with sustained virologic response 12 weeks post-treatment, defined as HCV RNA < LLOQ 12 weeks after the last dose of study drugs. Secondary endpoints included the percentage of patients with virologic failure during treatment (confirmed HCV RNA ≥ LLOQ after HCV RNA < LLOQ during treatment or confirmed HCV RNA ≥ LLOQ at the end of treatment) and the percentage of patients with post-treatment relapse (confirmed HCV RNA ≥ LLOQ between end of treatment and 12 weeks after the last dose of study drugs among patients completing treatment with HCV RNA < LLOQ at the end of treatment). Additional efficacy endpoints included percentage of patients with end of treatment response, defined as HCV RNA < LLOQ at week 12, sustained virologic response 4 weeks post-treatment, and sustained virologic response 24 weeks post-treatment. All analyses were performed on all patients who enrolled and received at least one dose of study drugs. SAS® (SAS Institute, Inc., Cary, NC) for the UNIX operating system was used for all analyses. All confidence intervals were 2-sided with an α level of 0.05.
From the intensive pharmacokinetic data, maximum observed plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) during a dosing interval (AUC12 for twice daily administration; AUC24 for once daily administration), and time to Cmax (Tmax) were summarized. Intensive pharmacokinetic data analyses were performed by non-compartmental methods using Phoenix® WinNonlin® Version 6.0 (Pharsight, A Certara Company, St. Louis, MO).
Results
Patients
Seventy-five patients were screened and 38 were enrolled. The most common reasons for exclusion were abnormal laboratory analysis results (16 patients, 7 of whom had hemoglobin less than the lower limit of normal), positive urine drug screen for barbiturates, amphetamines, cocaine, benzodiazepines, phencyclidine, propoxyphene, or alcohol (7 patients), and lack of verified infection with HCV genotype 1 (7 patients). Additional details of patients not meeting inclusion/exclusion criteria are in Supplementary Table 1. All enrolled patients received at least one dose of study drug (Fig. 1). The final date for data collection for analysis of sustained virologic response 12 weeks post-treatment was December 23, 2013. Patients were predominantly white (94.7%) and treatment-naïve (94.7%). Thirty-two of the 38 patients (84.2%) had HCV genotype 1a infection. Nineteen patients (50.0%) were on methadone and 19 (50.0%) were on buprenorphine. Baseline demographic and clinical characteristics of patients are shown in Table 1.
Efficacy
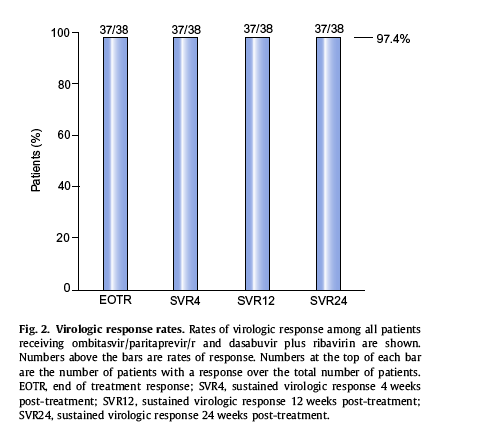
Thirty-seven of the 38 patients (97.4%) achieved sustained virologic response 12 weeks post-treatment. All of these patients also achieved end of treatment response and sustained virologic response 4 and 24 weeks post-treatment (Fig. 2). One patient (2.6%) prematurely discontinued the study after 25 days of treatment due to serious adverse events deemed by the investigator to be unrelated to study drug treatment. No patient had virologic failure during treatment or a post-treatment relapse.
Safety
The vast majority of patients (92.1%) experienced at least one adverse event (Table 2). Most of these events were mild or moderate in intensity, with only three patients (7.9%) experiencing a severe adverse event. The most common adverse events were nausea, fatigue, and headache. Two patients (5.3%) experienced serious adverse events, including one with cerebrovascular accident and sarcoma who withdrew from the study after 25 days of treatment. No other patients discontinued study drug due to adverse events. A second patient had a serious adverse event of acute myeloid leukemia reported 26 days post-treatment; this patient remained on study and achieved a sustained virologic response. None of these serious adverse events were considered related to study drug by the investigator.
Eight patients (21.1%) experienced a serum hemoglobin level <10 g/dl while on treatment, including two with a hemoglobin level <8.0 g/dl (grade 3). Ribavirin dosage was reduced for 6 of these patients, one of whom also received a blood transfusion. There were no virologic failures among these 6 patients, although one withdrew prematurely from the study as described above. One patient had a grade 3 elevation in total bilirubin (maximum value 5.8 mg/dl), which was predominantly of indirect bilirubin and improved with continued study drug administration. No patient discontinued study drug due to bilirubin elevation or other laboratory abnormalities. No patient required a change in the dosage of methadone or buprenorphine during study treatment.
Pharmacokinetics
A total of 22 of the 38 enrolled patients gave consent for intensive pharmacokinetic blood sampling. Twelve of these patients were on stable opioid replacement therapy of buprenorphine ± naloxone and 10 were on methadone. The steady-state pharmacokinetic parameters of dasabuvir, dasabuvir M1 metabolite, ombitasvir, paritaprevir, ritonavir, dasabuvir, and ribavirin are shown in Table 3. The steady-state exposures for ombitasvir, paritaprevir, and ritonavir in the present study were comparable to exposures observed in previous phase I studies of adults receiving similar formulations of ombitasvir/paritaprevir/ritonavir and dasabuvir without methadone or buprenorphine, while exposures of dasabuvir and dasabuvir M1 were slightly lower.
Discussion
Worldwide, HCV is a major cause of liver-related morbidity and mortality secondary to cirrhosis, end-stage liver disease, and hepatocellular carcinoma. Globally, 10 million PWID are estimated to be infected [2]; however, the majority of these patients remain untreated, with uptake rates for interferon-containing regimens of less than 10 percent and high rates of treatment discontinuation [[5], [6], [7], [12], [13]]. While lack of diagnosis and access to care are significant factors contributing to this low rate of treatment, the real and perceived side effects of interferon-based treatment also play a role [[21], [22]]. PWID may be dissuaded from seeking treatment based on information about the difficulties of interferon therapy obtained from peer networks [23]. The potential impact of interferon side effects on employment has also been identified as a barrier to treatment in this population [23]. In addition, PWID often have comorbidities such as depression, which make them poor candidates for interferon therapy.
Improved treatment uptake and completion rates could significantly decrease the burden of liver disease in this underserved patient population, since sustained virologic response is associated with a lower risk of HCV-related clinical outcomes [24]. Furthermore, although patients who have acquired HCV through a blood transfusion in the distant past are unlikely to transmit to others, those with a history of injection drug use remain a constant reservoir of potential transmission through relapse of substance abuse with unsafe injection practices. Mathematical models suggest that increased uptake of treatment among this patient population could substantially reduce HCV infection incidence and prevalence, leading to tangible benefits for overall public health [[25], [26]].
In this phase II, multicenter study of a 12-week regimen of ombitasvir/paritaprevir/r and dasabuvir plus ribavirin in HCV genotype 1 infected patients receiving opioid replacement therapy, 97.4% (37/38) of patients achieved sustained virologic response through 24 weeks post-treatment. No viral breakthroughs or relapses were observed with this multi-class approach to HCV treatment.
The virologic responses and low rate of discontinuation seen in this study were consistent with results from phase III trials evaluating this regimen, in which sustained virologic response rates of 96% were observed in treatment-naïve and peginterferon/ribavirin treatment-experienced patients who were not on opioid replacement therapy [[18], [20]]. Furthermore, low rates of reinfection among PWID who undergo successful HCV treatment (one to five percent per year) have been reported [25]. The phase II data reported here along with these low observed rates of reinfection suggest that a history of injection drug use should not be perceived as a barrier to HCV care and management by providers [[21], [25], [27], [28]].
In this study, ombitasvir/paritaprevir/r and dasabuvir plus ribavirin was generally well tolerated. Only one patient discontinued treatment prematurely, due to adverse events unrelated to study drug, and adverse events were generally mild or moderate. A grade 3 bilirubin elevation observed in one patient was predominantly indirect bilirubin, consistent with the known effect of paritaprevir on the bilirubin transporters OATP1B1 and OATP1B3 and the contribution of ribavirin and its associated hemolytic anemia [[29], [30]].
The frequency of hemoglobin values <10 g/dl was numerically greater in the current study compared to that seen in phase III trials of this regimen in genotype 1-infected patients who were not on opioid replacement therapy (21.1% vs. 5.1-5.8%) [[18], [20]]. This appears to be related to lower baseline hemoglobin levels in the current study compared to the previous studies (mean value 14.01 g/dl vs. 14.76-14.88 g/dl) since mean hemoglobin decreases were comparable (-2.09 g/dl vs. -2.33 to -2.52 g/dl). The frequency of ribavirin dose modification was correspondingly greater in the current study, but SVR12 rates were not affected by ribavirin dose modification in this study or in previous studies [[18], [20]].
Drug-drug interactions are a potential concern in patients on opioid replacement who are receiving direct-acting antiviral therapy. In this study, the high efficacy rates and low rates of discontinuation due to adverse events suggest that drug-drug interactions did not impact HCV treatment or opioid maintenance. The exposures of dasabuvir, dasabuvir M1 metabolite, ombitasvir, paritaprevir, and ritonavir in the current study were comparable to or slightly lower than the exposures observed in phase I studies of healthy volunteers receiving the HCV treatment alone. Drug-drug interaction studies in healthy volunteers not infected with HCV have also demonstrated a lack of effect of ombitasvir/paritaprevir/ritonavir and dasabuvir on methadone and buprenorphine ± naloxone as assessed by opioid pharmacodynamics, Pupillometry, Short Opiate Withdrawal Scale, and Desire for Drugs Questionnaire [[31], [32]]. While no data are available to assess the effect of ribavirin on buprenorphine ± naloxone or methadone, drug-drug interactions are not anticipated as ribavirin does not affect cytochrome P450-mediated metabolism and consequently does not interact with the metabolism of ombitasvir/paritaprevir/ritonavir and dasabuvir, or metabolism of opioid replacement therapies [[33], [34]]. The lack of observed drug-drug interactions in the current and previous studies suggests that no dose adjustment of methadone, buprenorphine ± naloxone, or HCV treatment is needed in patients on stable opioid replacement therapy and ombitasvir/paritaprevir/ritonavir and dasabuvir plus ribavirin.
Limitations of this study include the small sample size and selected patient population. Only patients receiving stable opioid substitution therapy were enrolled, effectively excluding those with active injection drug use. The latter is an important population that deserves further study. In addition, patients with cirrhosis were excluded from this study. However, 380 patients with cirrhosis were enrolled in a dedicated phase III trial of this treatment regimen, which demonstrated sustained virologic response rates of 92% and 96% following 12 or 24 weeks of treatment, respectively [35]. In addition, the current study was conducted prior to the availability of data from phase III trials evaluating the ombitasvir/paritaprevir/r and dasabuvir regimen ± ribavirin. Those trials demonstrated that genotype 1b-infected patients without cirrhosis treated with ombitasvir/paritaprevir/r and dasabuvir do not require ribavirin, since the regimen minus ribavirin achieved sustained virologic response rates of 100% in treatment-naïve and treatment-experienced genotype 1b-infected patients [[36], [37], [38]]. The efficacy of the ombitasvir/paritaprevir/r and dasabuvir regimen minus ribavirin in genotype 1b-infected patients receiving stable opioid substitution therapy was not evaluated in this trial.
With the low interferon-based treatment uptake rates observed in PWID [[5], [6], [7]], few HCV-infected individuals in this population achieve sustained virologic response. Opioid substitution therapy clinics may be an ideal setting to implement all-oral, interferon-free therapies for patients with HCV genotype 1 infection. These therapies have the potential to substantially limit disease progression and reduce transmission among patients with a history of injection drug use. The results of this study suggest that a 12-week regimen of ombitasvir/paritaprevir/r and dasabuvir plus ribavirin may be a suitable treatment option for this population. Given the burden of disease and pending availability of better tolerated and more efficacious regimens, greater efforts should be undertaken to screen, evaluate, and treat HCV-infected patients with a history of drug use [39].
|
|
| |
| |
|
|
|