| |
Sofosbuvir plus ribavirin for the treatment of chronic genotype 4
hepatitis C virus infection in patients of Egyptian ancestry
|
| |
| |
Download the PDF here
Journal of Hepatology May 2015
Peter J. Ruane1,, Dani Ain1, Richard Stryker1, Raymond Meshrekey1, Mina Soliman1,
Peter R. Wolfe1, Joseph Riad1, Sameh Mikhail1, Kathryn Kersey2, Deyuan Jiang2,
Benedetta Massetto2, Brian Doehle2, Brian J. Kirby2, Steven J. Knox2, John G. McHutchison2,
William T. Symonds2
1Ruane Medical and Liver Health Institute, Los Angeles, CA, USA; 2Gilead Sciences, Inc., Foster City, CA, USA
Background & Aims
We conducted an open-label phase 2 study to assess the efficacy and safety of the oral nucleotide polymerase inhibitor sofosbuvir in combination with ribavirin in patients of Egyptian ancestry, chronically infected with genotype 4 hepatitis C virus (HCV).
Methods
Treatment-naive and previously treated patients with genotype 4 HCV were randomly allocated in a 1:1 ratio to receive sofosbuvir 400 mg and weight-based ribavirin, for 12 or 24 weeks. The primary efficacy endpoint was the proportion of patients with sustained virologic response (HCV RNA <25 IU/ml) 12 weeks after cessation of therapy (SVR12).
Results
Thirty treatment-naive and thirty previously treated patients were enrolled and treated for 12 weeks (n = 31) or 24 weeks (n = 29). Overall, 23% of patients had cirrhosis and 38% had diabetes. 14% of treatment-naive patients were interferon ineligible and 63% of treatment-experienced patients had prior non-response. SVR12 was achieved by 68% of patients (95% CI, 49-83%) in the 12-week group, and by 93% of patients (95% CI, 77-99%) in the 24-week group. The most common adverse events were headache, insomnia, and fatigue. No patient discontinued treatment due to an adverse event.
Conclusions
The findings from the present study suggest that 24 weeks of sofosbuvir plus ribavirin is an efficacious and well tolerated treatment in patients with HCV genotype 4 infection.
Introduction
The genotype 4 strain of the hepatitis C virus (HCV) accounts for approximately 20% of all cases of chronic HCV infection worldwide [1]. In Egypt, where an estimated 15% of the population may have chronic hepatitis C, over 90% of the infections have been reported to be HCV genotype 4 [[1], [2], [3], [4]]. The spread of chronic HCV infection in Egypt is thought to be largely due to needle re-use during mass-treatment programs for schistosomiasis during the late 1950s through the early 1980s [[5], [6]]. Unfortunately, transmission continues to occur, primarily through iatrogenic sources, such as blood transfusions, injections, and dental care [[1], [2]]. HCV genotype 4 is also the most common genotype in other parts of the Middle East and Africa, and its prevalence is increasing in Europe and parts of North America where it has been associated with immigration and intravenous drug use [[1], [2]].
Until recently, the standard of care for genotype 4 HCV in the United States and Europe has been pegylated interferon (PegIFNα) with ribavirin (RBV) for 24 to 48 weeks, depending on virologic response [1]. Treatment-naive patients receiving this regimen have sustained virologic response (SVR) rates of 43% to 70% [[1], [2], [7]]. New regimens involving direct-acting antiviral agents (DAAs) have recently been approved for the treatment of genotype 4 HCV. These regimens appear to offer improved rates of SVR in treatment-naive and previously treated patients with genotype 4 HCV; however, few patients have received these regimens and data concerning efficacy and safety are sparse [8].
One of the newly approved DAAs indicated for the treatment of genotype 4 HCV is sofosbuvir (Gilead Sciences, Inc., Foster City, California, USA), an oral, HCV-specific NS5B nucleotide polymerase inhibitor with demonstrated clinical efficacy in patients with genotype 1 to 6 HCV [[9], [10]]. In a phase 3 trial, an SVR was observed in 27 of 28 treatment-naive patients (96%) with genotype 4 HCV receiving 12 weeks of sofosbuvir in combination with PegIFNα and RBV [9]. The current hepatitis C treatment guidelines for treatment of genotype 4 HCV issued by the American Association for the Study of Liver Diseases (AASLD), European Association for the Study of the Liver (EASL), and World Health Organization (WHO) include sofosbuvir administered in combination with PegIFNα and RBV for 12 weeks or an interferon-free regimen of sofosbuvir in combination with RBV for 24 weeks [[11], [12], [13]].
The development of an interferon-free regimen for genotype 4 HCV infection has the potential to significantly impact the incidence, prevalence, and overall burden of HCV, particularly in Egypt, where the prevalence of genotype 4 HCV is so high. For many, treatment with interferon-containing regimen is impossible, undesirable, or insufficiently efficacious. Elimination of interferon from the treatment regimen may reduce the required frequency of safety monitoring, and facilitate treatment of chronic hepatitis C in rural areas, which in Egypt have higher prevalence rates than the national average [4]. The large Egyptian immigrant population in the United States has afforded the opportunity to perform a pilot study of an interferon-free regimen containing sofosbuvir plus RBV in treatment-naive and treatment-experienced patients of Egyptian ancestry with HCV genotype 4 infection.
Patients and methods
Patients
Patients were screened and enrolled in this phase 2, open-label study at a single center in the United States between October 2012 and March 2013 (ClinicalTrials.gov Identifier NCT01713283). To be eligible, patients had to be born in Egypt to two parents of Egyptian ancestry. Patients were required to be at least 18 years of age with body mass index ≥18 kg/m2 and had chronic genotype 4 HCV infection with a serum HCV RNA level ≥104 log IU/ml. Up to 20% of enrolled patients may have had compensated cirrhosis at screening. The presence of cirrhosis was determined on the basis of a liver biopsy specimen showing evidence of cirrhosis or a Fibrotest® (Laboratory Corporation of America® Holdings, Burlington, North Carolina, USA) score >0.75 plus an aspartate aminotransferase:platelet ratio index (APRI) >2 during screening. Laboratory requirements included alanine and aspartate aminotransferase ≤10 times the upper limit of normal (ULN), direct bilirubin ≤1.5 times ULN, hemoglobin ≥12 g/dl for men and ≥11 g/dl for women, and creatinine clearance ≥60 ml/min (Cockcroft-Gault). Consistent with the inclusion of patients with cirrhosis, no minimum neutrophil count was required and patients with platelets >50,000/μl were eligible for participation. Patients with hepatitis B or HIV were excluded. Patients could be either treatment-naive or treatment-experienced; prior treatment with an anti-HCV direct-acting antiviral was exclusionary.
Hepatitis C virus GT 4 genotyping at screening was determined using the VERSANT® HCV Genotype 2.0 Assay (LiPA) (Siemens, Erlangen, Germany) or by population sequencing of a short fragment of the coding region of the NS5B gene (Janssen Diagnostics, Beerse, Belgium), subsequently confirmed by population or deep sequencing (DDL Diagnostic Laboratory, Rijswijk, The Netherlands). IL28B genotype was determined by means of polymerase chain-reaction amplification and sequencing of the rs12979860 single-nucleotide polymorphism.
The study was conducted in accordance with the guidelines of Good Clinical Practice and was approved by an Independent Review Board. All patients provided written informed consent.
Study design
Patients were randomly assigned in a 1:1 ratio to receive treatment with sofosbuvir plus RBV for 12 or 24 weeks. Randomization was stratified by prior treatment experience and cirrhosis status via a computer-generated random allocation sequence prepared by the sponsor. Sofosbuvir was given orally at a dose of 400 mg once daily, and weight-based RBV was given orally as a divided weight-based daily dose (1000 mg for patients with body weight <75 kg and 1200 mg with body weight ≥75 mg). RBV dose adjustment was permitted according to prescribing instructions. Use of growth factors was not permitted.
Efficacy assessments
Serum HCV RNA was measured using the COBAS® TaqMan® HCV Test v2.0 (Roche Molecular Systems, Pleasanton, California, USA; lower limit of quantitation (LLOQ) of 25 IU/ml) at baseline, all subsequent study visits during treatment, and post-treatment weeks 4, 12, and 24. Patients with confirmed HCV RNA < LLOQ at the end of treatment and post-treatment visits continued to the subsequent post-treatment visits, unless confirmed virologic relapse occurred. On-treatment virologic failure was defined as: breakthrough, i.e. confirmed HCV RNA ≥LLOQ after having previously had HCV RNA 1-log10 IU/ml increase in HCV RNA from nadir while on-treatment; or, non-response, i.e. HCV RNA persistently ≥LLOQ through 8 weeks of treatment. Relapse was defined as confirmed HCV RNA ≥LLOQ during the post-treatment period having achieved HCV RNA
Safety assessments
Safety was evaluated by assessment of clinical laboratory tests, physical examination, vital sign measurements, and documentation of adverse events. Safety data were collected from the first dose of study medication through 30 days after the last dose.
HCV resistance testing
For all patients who did not achieve SVR12, deep sequencing of the HCV NS5B gene, with a 1% assay cut-off, was performed at baseline and at the first virologic failure time point, if a serum sample was available and HCV RNA was >1000 IU/ml. The HCV NS5B coding region was amplified, and population- or deep-sequenced by DDL Diagnostic Laboratory (Rijswijk, The Netherlands) or Selah Genomics (Greenville, South Carolina, USA). Standard reverse transcription polymerase chain-reaction technology was used for amplification followed by deep sequencing of the polymerase chain-reaction product at baseline and virologic failure time points. Amino-acid substitutions in the NS5B coding region in the samples collected at virologic failure were compared with the respective baseline sequence for each patient. The availability of short fragment population sequence of the NS5B coding region from subtype determination allowed for baseline characterization of the S282 residue from all patients that achieved SVR12. NS5B nucleoside inhibitor (NI) RAVs were defined as the following substitutions at the following positions: S96T, N142T, L159F, S282any, M289I/L/V, L320F, and V321A.
Pharmacokinetics
Single blood samples were collected at baseline and each subsequent treatment visit for pharmacokinetic analysis. Plasma concentrations of sofosbuvir and its predominant circulating nucleoside metabolite (GS-331007) were determined using fully-validated high-performance liquid chromatography/tandem mass spectrometry bioanalytical methods (QPS, LLC, Newark, Delaware, USA). Plasma concentrations of sofosbuvir and GS-331007 were used to estimate pharmacokinetic exposure parameters (area under the concentration-time curve over the dosing interval [AUCtau] and the maximum concentration [Cmax]) for sofosbuvir and GS-331007 using population pharmacokinetics models previously developed with data from earlier phase 2/3 studies [14]. GS-331007 and sofosbuvir exposures observed in the patient population in this study were compared to exposures obtained in the phase 2/3 study population generated using the same population PK models.
Statistical assessments
The primary efficacy endpoint was the proportion of all randomized patients who achieved a sustained virologic response 12 weeks after the end of treatment (SVR12). Secondary efficacy endpoints included SVR4 and SVR24, on-treatment virologic failure, and virologic relapse after the end of treatment. In the primary efficacy analysis, SVR12 rates were calculated for each treatment group, along with 2-sided 95% confidence intervals (CIs) based on the Clopper-Pearson exact method. No statistical hypothesis testing was performed.
Results
Patients
Of the 80 patients screened, 60 patients were enrolled and completed treatment with sofosbuvir plus RBV for 12 weeks (n = 31) or 24 weeks (n = 29) (Fig. 1).
The 12- and 24-week treatment groups were balanced for demographic and baseline characteristics, except that the 12-week group had a greater proportion of patients with HCV genotype 4a (Table 1). The mean age of enrolled patients was 54 years, 68% were men, 80% had genotype 4a infection, 83% had IL28B non-CC genotype, 33% had a body mass index ≥30 kg/m2, 23% had cirrhosis, 38% had diabetes, and all but two had platelet counts >100,000/l. Of the 14 patients with protocol-defined cirrhosis, 11 had a historical or screening liver biopsy specimen showing evidence of cirrhosis; the other three were judged to have cirrhosis on the basis of having a Fibrotest score >0.75 plus an APRI score >2. Among treatment-naive patients, 14% were interferon ineligible. Among treatment-experienced patients, the most common reason for prior HCV treatment failure was non-response, occurring in 63% of patients.
Efficacy
HCV RNA levels declined rapidly upon initiation of treatment, from a mean of 5.98 log10 IU/ml at baseline to 1.81 log10 IU/ml in the 12-week group and from 5.97 log10 IU/ml to 1.74 log10 IU/ml in the 24-week group after one week of treatment. All patients except one in the 12-week group had HCV RNA below the lower limit of quantification by week 4.
SVR12 was achieved by 21 of the 31 patients (68%; 95%, CI 49-83%) receiving 12 weeks of treatment, and by 27 of the 29 patients (93%; 95%, CI 77-99%) receiving 24 weeks of treatment (Table 2). In general, differences in favor of 24 weeks of treatment were observed for most patient subgroups, although the numbers of patients within each subgroup were often small. SVR12 rates were numerically higher in patients younger than 65 years and in patients with body mass index ≥30 kg/m2 in both the 12 and 24-week treatment groups, while numeric differences were seen in the 12-week but not the 24-week group for those with cirrhosis, high baseline viral load, or IL28B non-CC genotype. The SVR12 rates among treatment-naive patients were higher (79% in the 12-week group and 100% in the 24-week group) than among treatment-experienced patients (Table 2). The rates of SVR12 in patients with genotype 4a vs. non-4a infection were similar: 81% vs. 75%. RBV dose reduction did not appear to have an effect on SVR12 rate: of the 11 patients who received RBV dose reduction during treatment, 9 (82%) achieved SVR12.
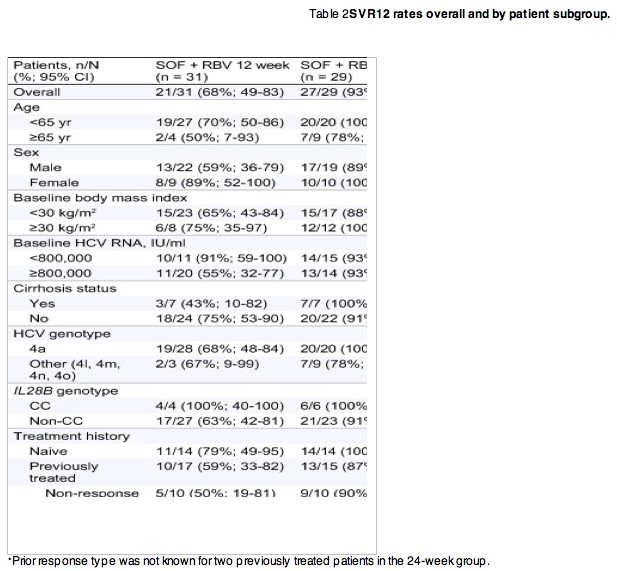
In total, 10 patients in the 12-week group and 2 patients in the 24-week group had virologic failure. Ten of the 12 patients relapsed by week 4 post-treatment and one patient in the 24-week group relapsed between week 4 and week 12 post-treatment. One treatment-naive patient in the 12-week group had on-treatment virologic failure (breakthrough); this 64-year-old female patient had genotype 4l HCV, cirrhosis, and the IL28B CT genotype. She completed 12 weeks of treatment and achieved HCV RNA < LLOQ by week 8, but had quantifiable HCV RNA at treatment weeks 10 and 12. All patients with SVR12 who returned for the 24-week post-treatment visit achieved SVR24; two were lost to follow-up.
Viral sequencing
Viral isolates from all 60 patients enrolled had sequencing performed of the region covering the sofosbuvir resistance-associated variant site S282. No variants in this region were observed at baseline in the 49 patients with population sequencing for this region or in the 11 with full NS5B deep sequencing data. No S282T variants were observed at the virologic failure time points in the 12 patients who did not achieve SVR12, including the patient who had viral breakthrough on-treatment. One patient who relapsed harbored a viral population with a low level (1.6%) of the S282G variant; the clinical significance of this variant is unclear as this mutant does not replicate in vitro. No other sofosbuvir treatment-emergent variants were observed in the 11 samples successfully deep-sequenced.
Pharmacokinetics
GS-331007 and sofosbuvir population pharmacokinetic model-derived exposure parameters AUCtau and Cmax were generated for all patients with measureable plasma concentrations of GS-331007 or sofosbuvir. Mean exposure of GS-331007, the renally eliminated metabolite of sofosbuvir, was moderately higher (AUCtau 46%, Cmax 42%) in this study population than in HCV-infected patients in earlier phase 2/3 studies, even though CLcr values were comparable in the two populations [14]. The mechanism for this modest increase is unknown. The increased GS-331007 exposure was not clinically relevant as the range of exposures observed in the Egyptian population was comparable to that observed in previous studies in which there had been no exposure-related toxicities. Mean exposure of sofosbuvir was comparable in the two populations. No clinically relevant effects of age, sex, body mass index, creatinine clearance, or cirrhosis on GS-331007 or sofosbuvir exposure were observed, which is consistent with findings from previous studies. No clinically relevant differences in GS-331007 or sofosbuvir exposure were observed for study patients who received 12 vs. 24 weeks of sofosbuvir plus RBV, or in treatment-naive vs. treatment-experienced patients, or in those who achieved SVR12 vs. those with virologic failure.
Safety
Treatment-emergent adverse events, which were reported in 28 patients (90%) in the 12-week group and 29 (100%) in the 24-week group, were predominantly mild or moderate in severity. The most common adverse events in both groups were headache, insomnia, and fatigue (Table 3). Although some AEs were more common in the 24-week group, this does not appear to be the result simply of longer duration, since the differences between the arms were observed during the first 12 weeks of treatment.
Serious adverse events were reported in three patients in the sofosbuvir plus RBV 24-week group. One patient experienced severe non-cardiac chest pain and a second patient had severe abdominal pain during the dosing period. The third patient experienced transient loss of consciousness after a transatlantic flight four days after completing treatment. All these adverse events resolved and none was considered to be treatment related.
Consistent with changes in laboratory values typically associated with RBV, decreases from baseline in hemoglobin and increases in reticulocytes and platelets were observed during treatment. Four patients, all in the 24-week sofosbuvir plus RBV group, had at least one hemoglobin level of <10 g/dl, but none had hemoglobin <8.5 g/dl. Consistent with RBV-induced hemolysis, one patient in each of the 12 and 24-week groups had a total bilirubin value >2.5 times the upper limit of the reference range at week 1; values at subsequent visits decreased during continued treatment. Serum glucose values >250 mg/dl occurred in six patients in the 12-week sofosbuvir plus RBV group and three patients in the 24-week group, all of whom had concomitant diabetes.
Adverse events resulted in dose modification or interruption of RBV in two patients (6%) in the sofosbuvir plus RBV 12-week group and 9 (31%) in the 24-week group. Moderate dyspnea led to permanent discontinuation of RBV on day 36 of treatment in a treatment-experienced patient in the 24-week group; sofosbuvir was continued for the remainder of the treatment period and the patient achieved SVR24. No patients had adverse events leading to dose modification, interruption, or discontinuation of sofosbuvir.
Discussion
In this phase 2, open-label study, 24 weeks of treatment with sofosbuvir and RBV resulted in high rates of SVR12 in treatment-naive and previously treated patients with genotype 4 HCV. SVR12 rates were notably high in patients with characteristics historically associated with poor response-cirrhosis, high baseline viral load, non-CC IL28B genotype, and prior non-response to HCV treatment. The regimen was well tolerated, with mostly mild adverse events typically associated with RBV therapy. Overall, RBV dose modification or interruption did not appear to have an effect on SVR. No viable resistance-associated variants were detected in any of the patients who did not achieve SVR.
Overall and in nearly every patient subgroup, patients receiving 24 weeks of treatment had substantially higher rates of SVR12 than patients receiving 12 weeks of treatment. In some subgroups - e.g., males, cirrhotics, patients with genotype 4a HCV - the difference was 30 percentage points or more.
Unsurprisingly, treatment-naive patients overall had higher rates of SVR12 than did treatment-experienced patients, but among treatment-naive patients, the SVR rate was 21 percentage points higher among those receiving 24 weeks of treatment than those receiving 12 weeks (Table 4). On the basis of these preliminary data, the only subgroups for whom 12 weeks of this regimen would appear to be adequate are patients with low baseline HCV RNA levels and those with the CC IL28B genotype. However, any such conclusions are necessarily preliminary given the small numbers of patients in some subgroups.
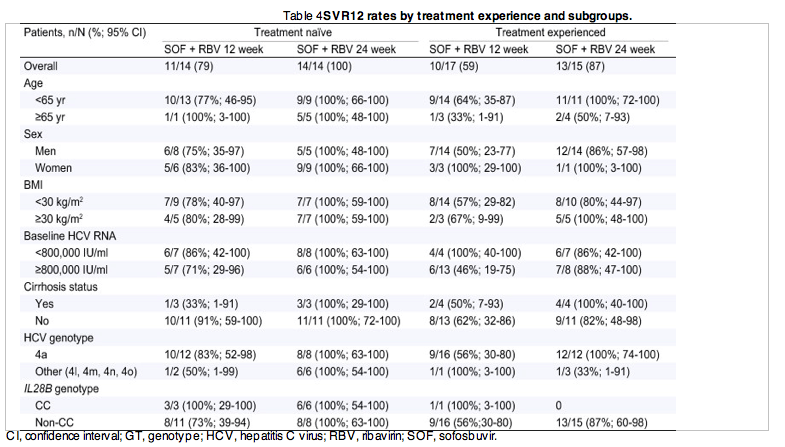
The results in our 24-week arm compare favorably with other recently approved regimens for which data are currently available in patients with genotype 4 HCV. In a phase 3 trial, the second generation HCV NS3/4A protease inhibitor simeprevir was administered for 12 weeks with PegIFNα and RBV followed by a further 12-36 weeks of PegIFNα and RBV (depending on on-treatment response) to 107 patients with genotype 4 HCV. The overall rate of SVR12 was 65%, but the rate varied greatly by treatment history: 83% in treatment-naive patients, 86% in prior relapsers, 60% in prior partial responders, but only 40% in patients with prior non-response [15]. Although results above are comparable to our results in the 12-week arm for treatment-naive and previous relapsers, treatment with sofosbuvir plus RBV appears to be better tolerated. Simeprevir administered in combination with sofosbuvir is also approved in Europe for the treatment of genotype 4 patients who are interferon-intolerant or ineligible, but no data are presently available for this regimen in patients with genotype 4 HCV.
Other direct-acting antiviral agents have also been evaluated in phase 2 studies in patients with genotype 4 HCV infection. One such study evaluated 12 weeks of treatment with the protease inhibitor ABT-450 with ritonavir (ABT-450/r) and the NS5A inhibitor ombitasvir, with or without RBV, in genotype 4 patients without cirrhosis [16]. In treatment-naive patients, the RBV-containing regimen resulted in a 100% SVR12 rate (n = 42/42), while the regimen without RBV resulted in a 91% SVR12 rate (n = 40/44). The SVR12 rate has not yet been reported for the group of treatment-experienced patients (n = 49) who received ABT-450/r plus ombitasvir plus RBV. In a small study, a total of 21 treatment-naive genotype 4 patients were randomized to receive daclatasvir, a NS5A inhibitor, and asunaprevir, a NS3 protease inhibitor, and one of two dose levels of BMS-791325, a non-nucleoside NS5B polymerase inhibitor, for 12 weeks [17]. All 21 patients achieved SVR12, suggesting the combination of these agents merits further evaluation. The safety and efficacy of sofosbuvir in combination with the NS5A inhibitor ledipasvir in patients with genotype 4 is being evaluated in studies in Egypt and France.
There is a need in Egypt for an interferon-free regimen that is well tolerated and provides a high degree of efficacy for the treatment of genotype 4 HCV infection. However, the results from this study may have broader application. Although some studies have found response rates with interferon plus RBV to be higher in Egyptian patients than European patients with genotype 4 HCV infection [[18], [19]], other studies have not found a difference in response based on ethnicity [[20], [21]]. Differences in efficacy have been associated with differences in patient characteristics including genotype 4a, which predominates in Egypt whereas in Europe genotypes 4a and 4d are common and greater subtype diversity is present in patients from Africa [[20], [22]]. The IL28B CC genotype has been associated with higher response rates to treatment with interferon plus RBV in genotype 4 HCV infection [23] and, in turn, a higher frequency of the C allele was found in Egyptian patients relative to Europeans and Sub-Saharan Africans [23]. In this study, 20% of patients had a subtype other than genotype 4a and 83% had IL28B non-CC status. Regardless of genotype 4 subtype or IL28B status, sofosbuvir plus RBV administered for 24 weeks resulted in high SVR12 rates.
Limitations of this study include the absence of complete prior treatment histories in all patients, particularly those who had previously been treated for HCV in Egypt, and the small number of patients infected with non-4a HCV. The absence of data in patients with other genotype 4 subtypes and other racial and ethnic groups does not permit us to speculate about the benefit of the regimen for these populations.
Effective interferon-free regimens are associated with important advantages in treating chronic HCV, including sparing patients the rigors and toxicity of protracted interferon therapy. The increasing availability of such regimens has spurred calls for stepped up screening for HCV in countries of high endemicity [24]. The findings from the present study suggest that sofosbuvir plus RBV may offer an efficacious and well tolerated treatment in patients with HCV genotype 4 infection, and one that may facilitate treatment of large numbers of Egyptian patients. A phase 3 trial of sofosbuvir plus RBV for 12 or 24 weeks in patients with genotype 4 is ongoing in Egypt.
|
|
| |
| |
|
|
|