| |
Ledipasvir and sofosbuvir for hepatitis C genotype 4:
a proof-of-concept, single-centre, open-label phase 2a cohort study
|
| |
| |
Download the PDF here
Download the PDF here
Lancet Infectious Diseases July 2015
Anita Kohli, Rama Kapoor, Zayani Sims, Amy Nelson, Sreetha Sidharthan, Brian Lam, Rachel Silk, Colleen Kotb, Chloe Gross, Gebeyehu Teferi,
Kate Sugarman, Phillip S Pang, Anu Osinusi, Michael A Polis, Vinod Rustgi, Henry Masur, Shyam Kottilil
Summary
Background
Worldwide, although predominantly in low-income countries in the Middle East and Africa, up to 13% of hepatitis C virus (HCV) infections are caused by HCV genotype 4. For patients with HCV genotype 1, the combination of ledipasvir and sofosbuvir has been shown to cure high proportions of patients with excellent tolerability, but this regimen has not been assessed for the treatment of HCV genotype 4. We assessed the efficacy, safety, and tolerability of 12 weeks of combination therapy with ledipasvir and sofosbuvir for patients with chronic HCV genotype 4 infections.
Methods
In this single-centre, open-label cohort, phase 2a trial, patients with HCV genotype 4 who were treatment naive or interferon treatment experienced (HIV-negative) were sequentially enrolled at the Clinical Center of the National Institutes of Health, Bethesda, MD, USA. We gave patients 12 weeks of ledipasvir (90 mg) and sofosbuvir (400 mg) as a single combination tablet once per day. The primary efficacy endpoint was sustained viral response at 12 weeks (SVR12), as measured by the proportion of patients with HCV RNA concentrations less than the lower limit of quantification (COBAS TaqMan HCV test, version 1.0, 43 IU/mL). The primary safety endpoint was the frequency and severity of adverse events. We did our analyses on an intention-to-treat basis. This study is registered with ClinicalTrials.gov, number NCT01805882.
Findings
Between Sept 16, 2013, and Nov 2, 2014, we recruited 21 patients. 20 (95%) of 21 patients completed 12 weeks of treatment and achieved SVR12 (95% CI 76-100), including seven patients with cirrhosis. One patient was non-adherent to study drugs and withdrew from the study, but was included in the intention-to-treat analysis. No patients discontinued treatment because of adverse events and no grade 3 or 4 adverse events occurred that were related to study medications. The most common adverse events were diarrhoea (two patients), fatigue (three patients), nausea (two patients), and upper respiratory infections (two patients).
Interpretation
Ledipasvir and sofosbuvir treatment for 12 weeks was well tolerated by patients with HCV genotype 4 and resulted in 100% SVR for all patients who received all 12 weeks of study drugs, irrespective of previous treatment status and underlying liver fibrosis. This is the first report of a single-pill, all-oral, interferon-free, ribavirin-free treatment for patients with HCV genotype 4.
Funding
NIAID, National Cancer Institute and Clinical Center Intramural Program. The study was also supported in part by a Cooperative Research and Development Agreement between NIH and Gilead Sciences.
Introduction
Roughly 185 million people worldwide are infected with hepatitis C virus (HCV), which is associated with progression to end-stage liver disease and hepatocellular carcinoma.1, 2 HCV genotype 4 accounts for about 8-13% of these infections, mainly concentrated in sub-Saharan Africa, northern Africa, the Middle East, and southeast Asia.3, 4 Within Europe, HCV genotype 4 is present in a substantial proportion of patients with HCV infections in countries including Belgium, France, and Greece.5 In Egypt, genotype 4 HCV is particularly common, and about 15% of the Egyptian population is infected with this subtype.3
Although the development of ribavirin-free and interferon-free regimens for HCV genotype 1 has progressed rapidly, simple well tolerated treatments for HCV genotype 4 are particularly crucial in view of the high concentration of this genotype in low-income countries, where laboratory monitoring for adverse events might not be feasible.4 In 2014, the directly acting antivirals sofosbuvir and simeprevir were introduced. Sofosbuvir or simeprevir in combination with pegylated interferon and ribavirin for 12 weeks substantially improved the proportion of patients with HCV genotype 4 achieving a sustained viral response (SVR).6 For patients who were ineligible for interferon treatment, sofosbuvir and ribavirin for 24 weeks was also recommended.6 Although these regimens are associated with high 59-100% SVR for patients who are treatment naive or treatment experienced, they include multiple pills, injections, and long treatment durations.6Additionally, both interferon and ribavirin need frequent laboratory monitoring and are associated with toxic effects, including fatigue, anaemia, and teratogenicity.6
Research in context
Systematic review
We searched PubMed on Feb 11, 2015, for articles published between Jan 1, 2000, and Feb 11, 2015, using a combination of the MeSH search terms �HCV treatment�, �antiviral agent�, and �genotype 4� and consulted the hepatitis C virus (HCV) treatment guidelines for phase 2 or 3 clinical trials of treatments for patients with HCV genotype 4. We also searched the reference lists of articles from our search for additional reports that met our inclusion criteria of phase 2 and phase 3 clinical trials of interferon-free regimens for treatment of HCV genotype 4.
Four clinical trials have been reported (one journal article and four in abstract form) for interferon-free regimens for patients with HCV genotype 4. The results of these trials have shown promising safety and efficacy (sustained viral response at 12 weeks, 84-100%) with combination direct-acting antiviral drugs, with or without ribavirin for 12-24 weeks. Few patients with cirrhosis or who have previously been treated with interferon-containing regimens have been included.
Added value of this study
Although our study is small, we showed high rates of sustained viral response at 12 weeks with use of sofosbuvir and ledipasvir for 12 weeks, which supports the possibility that this simple regimen might be effective for some patients.
Implications of all the available evidence
Further development of this efficacious, simple, well tolerated regimen is warranted and studies in patients with cirrhosis and previously treated patients should be pursued.
Two directly acting antivirals, ledipasvir (an NS5A inhibitor) and sofosbuvir (a nucleotide polymerase inhibitor), have been approved in the USA for combination use for the treatment of HCV genotype 1. Results from studies of this combination, given as one pill per day for 12 weeks, have shown 91-100% SVR in patients with HCV genotype 1 who are treatment naive and treatment experienced, with few adverse events.7, 8 In vitro, both ledipasvir and sofosbuvir have anti-HCV activity against HCV genotype 4 that is similar to that noted against HCV genotype 1. However, the clinical efficacy of this regimen in vivo for HCV genotype 4 has not yet been established.
Historically, proportions of patients with HCV genotype 4 who achieve SVR when given interferon-containing treatments have been between those of patients with HCV genotype 1 and HCV genotypes 2 and 3.9, 10 Furthermore, the in-vitro efficacy of ledipasvir and sofosbuvir for HCV genotype 4 suggests that the combination of these potent directly acting antivirals without ribavirin for 12 weeks might be effective for the treatment of HCV genotype 4 in patients who are treatment naive or interferon treatment experienced. Therefore, we did a clinical trial to assess the two drug combination of ledipasvir and sofosbuvir for 12 weeks for the treatment of patients with HCV genotype 4.
Methods
Study design
In this single-centre, open-label cohort, non-randomised phase 2a trial, we sequentially enrolled patients with HCV genotype 4 at the Clinical Center of the National Institutes of Health, Bethesda, MD, USA. The study was approved by the Institutional Review Board of the National Institute of Allergy and Infectious Diseases (NIAID) and was done in compliance with the Good Clinical Practice guidelines, the Declaration of Helsinki, and regulatory requirements. The Regulatory Compliance and Human Participants Protection Branch of NIAID served as the study sponsor and medical monitor. Gilead Sciences provided drug and scientific advice.
Patients
Eligible participants were men and women, aged 18 years or older, with chronic HCV genotype 4 infection (serum HCV RNA �2000 IU/mL) and compensated liver disease, and who were treatment naive or treatment experienced (previous exposure to directly acting antivirals was exclusionary). We excluded patients with HIV or hepatitis B virus infection. We invited all eligible patients at the study centre to enrol. We contacted patients for screening visits in the order in which they initially contacted the study team for participation and enrolled them in the order in which they completed screening requirements. We measured the stage of liver disease by liver biopsy or a Fibrosure test. We did not use Fibroscan because of its low availability at most medical care centres in the USA at the time of study initiation. The appendix includes full eligibility criteria. We obtained written or oral informed consent from all participants.
Procedures
We gave patients 90 mg ledipasvir and 400 mg sofosbuvir as a single combination tablet once per day for 12 weeks. We stopped giving study drugs if patients did not achieve more than a 2 log10 reduction in HCV RNA by week 4, unless a 2 log10 or more reduction would be less than the lower limit of HCV quantification. We included all patients who took at least one pill in the final analysis. In accordance with the protocol, if participants failed treatment, they were offered treatment with the standard of care, which at the time of the study was pegylated interferon and ribavirin or ribavirin alone with sofosbuvir. Neither patients nor investigators were masked to treatment allocation.
We measured plasma HCV RNA concentrations using the RealTime HCV Assay (Abbott, Chicago, IL, USA), with a lower limit of quantification of 12 IU/mL and a lower limit of detection of 3 IU/mL, at the start of the study; weeks 4, 8, and 12 of treatment; and 2, 4, 8, and 12 weeks after treatment. We also measured serum HCV RNA levels with the COBAS TaqMan HCV RNA assay, version 1.0 (Roche, Pleasanton, CA, USA), with a lower limit of quantification of 43 IU/mL and a lower limit of detection of 15 IU/mL at screening, day 0 and weeks 4, 8, 12, 36, 48, and 60.
We recorded adverse events and clinical laboratory results throughout the study. We graded adverse events from grade 1 (mild) to grade 4 (severe) in accordance with the NIAID Division of AIDS (DAIDS) toxicity table (version 1.0).11
Outcomes
The primary efficacy outcome was SVR12, defined as the proportion of participants with plasma HCV viral load less than the lower limit of quantification of the Roche COBAS TaqMan HCV RNA assay 12 weeks after treatment completion. The lower limit of quantification is the lowest HCV RNA concentration that is within the linear range of detection of the HCV RNA assay.
The primary safety outcome was the frequency and severity of adverse events. Secondary outcomes that we completed and included in this study are the proportion of participants with unquantifiable HCV viral load at specified timepoints during and after treatment, treatment discontinuations, adverse events, and safety laboratory changes. Data up to and including SVR12 are reported in this study and follow-up for 48 weeks post-treatment is ongoing.
Statistical analysis
The primary efficacy and safety analyses were based on an intention-to-treat population (all patients who received at least one dose of study drugs). We calculated the sample size to provide both a sufficiently high probability of identification of at least one adverse event of probability 10% or more and with prespecified CIs for estimates of efficacy, assuming 20 patients. Because one patient dropped out at week 5 and was replaced, we calculated post-hoc CIs for 21 patients. With 21 patients in the treatment group, if the true probability of an adverse event caused by a regimen is 10% or more, a sample size of 21 provides an 88% chance of identification of at least one such adverse event. With a sample size of 21, if all patients achieved SVR12, the 95% CI for that estimate is 87-100 and if 19 patients achieved SVR12, the 95% CI for that estimate would be 76-100. We calculated the proportion of patients achieving SVR12 after completion of therapy. We did analyses with PRISM 6.0. This study is registered with ClinicalTrials.gov, number NCT01805882.
Role of the funding source
Data collection, review, and analysis were done by NIH and University of Maryland investigators. AK and SK participated in the study design and all authors contributed to the writing of the report. NIH and University of Maryland-affiliated investigators (AK, RK, ZS, AN, SS, RS, CK, CG, MAP, HM, and SK) had full access to all data in the study, and AK and the corresponding author had final responsibility for the decision to submit for publication.
The Regulatory Compliance and Human Participants Protection Branch of the National Institute of Allergy and Infectious Diseases (NIAID) served as the study sponsor and were involved in the review and approval of the study via the usual peer-review process as well as the study management. They did not have a role in the design of the study, data collection and analysis, interpretation of the data, preparation, review, or approval of the manuscript, or the decision to submit the manuscript for publication.
Results
We enrolled and followed up patients from Sept 16, 2013, to Nov 2, 2014. We screened 24 participants and 21 were enrolled in study (figure). Table 1 shows baseline characteristics of the study population.
20 (95%, 95% CI 76-100) of 21 patients treated with ledipasvir and sofosbuvir achieved SVR12, in accordance with our predefined criteria. One patient had an HCV RNA concentration of 1 533 291 IU/mL at week 4. On further questioning the patient reported taking only one dose of study drug by week 4. This patient withdrew from the study at week 5, but is included in the intention-to-treat analysis. In accordance with the protocol, we replaced this patient, but included all 21 patients in the analysis.
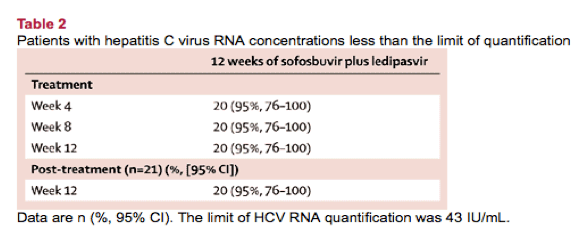
20 (95%) of 21 participants treated with ledipasvir and sofosbuvir had concentrations of HCV RNA that were less than the lower limit of quantification by both week 4 and week 8 of treatment (table 2). The appendix contains complete results for HCV RNA and changes in alanine transaminase and aspartate aminotransferase.
20 (95%) of 21 patients completed treatment. The most common adverse events were diarrhoea, fatigue, nausea, and upper respiratory infections (table 3). Most adverse events were mild in severity. No deaths, serious adverse events, or grade 3 or 4 adverse events occurred. We identified five grade 3 laboratory abnormalities: decreased absolute neutrophil count in a patient who reported taking only one dose of study medication 4 weeks before we detected this effect; hyperglycaemia in two patients with type 2 diabetes mellitus and haemoglobin A1C fractions of 7·6% and 8·7%, respectively, at screening; hypophosphataemia in a patient with a history of grade 3 hypophosphataemia before starting study drugs; and thrombocytopenia (41 000 cells per mL) in a patient with cirrhosis and thrombocytopenia (45 000 cells per mL) before starting study drugs.
Discussion
In the present study, treatment with sofosbuvir and ledipasvir for 12 weeks resulted in 95% SVR12 in patients with chronic HCV genotype 4 infections who were treatment naive or interferon treatment experienced. The regimen was well tolerated, rapidly suppressed HCV viraemia, and substantially simplified treatment for HCV genotype 4. 100% of patients who received the full 12 weeks of therapy achieved SVR12.
Treatment for HCV infection is changing rapidly.9 Results from early studies showed that sofosbuvir or simeprevir could be used in combination with pegylated interferon and ribavirin for the treatment of HCV genotype 4 infection.
Subsequently, sofosbuvir and ribavirin alone for 12-24 weeks was shown to result in 59-100% SVR in patients with HCV genotype 4 who were treatment naive and treatment experienced,14 with similar results in HIV/HCV genotype-4-co-infected patients.15 In the PEARL-1 trial,16 paritaprevir and ombitasvir, with or without ribavirin, were assessed in patients with HCV genotype 4 who were treatment naive, and with ribavirin in patients who previously did not respond to interferon treatment. This regimen resulted in SVR12 in 42 (100%)of 42 patients who were treatment naive and received ribavirin, 40 (91%) of 44 patients who were treatment naive and did not receive ribavirin, and 49 (100%) of 49 in those who were treatment experienced.16
Although many interferon-free and ribavirin-free regimens are in development for HCV genotype 1, development of interferon-free and ribavirin-free regimens for HCV genotype 4 has been slow. In our proof-of-concept study, the use of ledipasvir and sofosbuvir to treat HCV genotype 4 in mainly immigrant patients who were treatment naive and treatment experienced resulted in a high proportion of patients being cured, similar to or exceeding those of the standard of care,14, 16, 17 with excellent tolerability. More than half of patients had a high baseline viral load, were treatment experienced, or had advanced stage 3 or 4 liver disease-factors previously associated with treatment failure in HCV genotype 1 patients.18, 19 Despite lower EC50 values in in-vitro replicons for ledipasvir against HCV genotype 4 (0·39 nM for genotype 4 vs 0·031 nM against genotype 1a and 0·004 nM against genotype 1b), 100% of patients who completed therapy achieved SVR, thus supporting the potency of NS5A inhibitors.20
Although 12 weeks of therapy was effective in all patients who completed it, one patient in this trial was non-adherent to the study drugs and discontinued the study at week 7. The patient was treatment naive and had stage 1-2 liver disease.
Pending validation by larger trials, the results of our study suggest that the use of 12 weeks of ledipasvir and sofosbuvir is effective for the treatment of HCV genotype 4. A safe and simple, single-pill regimen will be ideal to treat large numbers of patients and therefore affect the global hepatitis genotype 4 epidemic. Whether therapeutic regimens could be shortened further for patients with specific host or viral parameters and comorbidities remains to be established. Further reductions in treatment duration could decrease the costs of treatment, which are high.21 Additionally, licensing agreements in some low-income countries might also allow for further reductions in drug prices.22
Limitations of the study include the sequential, non-randomised enrolment, which we chose because of the toxic effects and low SVR associated with the standard of care at time of study initiation (pegylated interferon and ribavirin),23 which would have been the comparator arm. Additionally, this was a single-site trial, which we deemed to be sufficient in view of the exploratory nature of this study. Confidence in the estimates of efficacy is limited by the small numbers of patients included and the ability to use only historical comparisons for efficacy. We included patients with and without cirrhosis and those who were treatment naive and treatment experienced, but the study was not powered to compare these groups. Most patients had early stage liver fibrosis (less than stage 3); other studies suggest that these patients might have more favourable responses to directly acting antiviral treatment compared with patients with advanced liver fibrosis (stage 3 or cirrhosis).19 Further studies in patients with advanced stage 3 or 4 liver disease should be done. Although IL28B genotype has not been shown to be a useful predictor of treatment outcome for sofosbuvir and ledipasvir when used to treat HCV genotype 1,8 we did not examine this factor in our study of HCV genotype 4. Additionally, patients received intensive nursing support and monitoring, which might not be replicable in community-based treatment programmes for hepatitis C.
In conclusion, 12 week regimens of oral combination directly acting antiviral treatments with ledipasvir and sofosbuvir seem to be effective in the treatment of patients with HCV genotype 4. This simple, well tolerated treatment for HCV genotype 4 holds promise to substantially improve and simplify the treatment of HCV in the low-resource countries where this virus genotype is concentrated.
Contributors
SK and AK had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. AK and ZS did the literature search. AK, SK, AN, MAP, and HM contributed to the study design. AK, ZS, RK, SS, AN, CK, CG, GT, KS, and SK collected the data. AK, ZS, RK, SS, and MAP analysed the data. AK, AO, SK, RK, PSP, GT, KS, VR, MAP, BL, and HM interpreted data. ZS, AK, and RK contributed to figure design. AK wrote the first draft of the manuscript and all authors participated in the review and critiquing of the manuscript.
Declaration of interests
AO and PSP are employees of Gilead Pharmaceuticals. GT serves on the Gilead and Merck advisory boards and as a speaker for Gilead. All other authors declare no competing interests. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organisations imply endorsement by the US Government.
Acknowledgments
This project was funded with federal funds from the National Cancer Institute of the National Institutes of Health (NIH), under Contract No. HHSN261200800001E. This research was supported in part by the National Institute of Allergy and Infectious Diseases and in part by a Cooperative Research and Development Agreement between NIH and Gilead Sciences. These entities did not have a role in the writing of the manuscript or decision to submit for publication. We would like to acknowledge the contributions of the following individuals: Katie Watkins, Erin Rudzinski, and Susan Vogel (clinical monitoring support); Judith Starling and Lori Gordon (pharmacy); Vanessa Eccard, Jerome Pierson, and John Tierney (regulatory support); William Ronnenberg, Richard Williams, and Mike Mowatt (technology transfer support); Marc Teitelbaum (sponsor medical monitor); John Powers (oversight); Mary Hall (protocol support); Cathy Rehm, Sarah Jones, and Kerry Townsend (clinical support); and Senora Mitchell (clinic support).
|
|
| |
| |
|
|
|