| |
HIV-1 Eradication: Early Trials (and Tribulations)
|
| |
| |
Download the PDF here
Adam M. Spivak1 and Vicente Planelles2,*
1Department of Medicine, University of Utah School of Medicine, Salt Lake City, UT, USA 2Department of Pathology, University of Utah School of Medicine, Salt Lake City, UT, USA
Antiretroviral therapy (ART) has rendered HIV-1 infection a manageable illness for those with access to treatment. However, ART does not lead to viral eradication owing to the persistence of replication-competent, unexpressed proviruses in long-lived cellular reservoirs. The potential for long-term drug toxicities and the lack of access to ART for most people living with HIV-1 infection have fueled scientific interest in understanding the nature of this latent reservoir. Exploration of HIV-1 persistence at the cellular and molecular level in resting memory CD4+ T cells, the predominant viral reservoir in patients on ART, has uncovered potential strategies to reverse latency. We review recent advances in pharmacologically based 'shock and kill' HIV-1 eradication strategies, including comparative analysis of early clinical trials.
Trends
ART blocks ongoing viral replication but does not cure HIV-1 infection owing to the presence of long-lived quiescent proviruses in a minority of resting CD4+ T cells (known as the latent reservoir).
ART administration significantly lowers the risk of viral transmission, and therefore serves as individual treatment and public health prevention. However, worldwide incident infections continue to outpace the number of people started and maintained on ART. Global resource limitations that contribute to this imbalance suggest that ART alone will not be enough to curb the HIV-1 epidemic.
The Successes and Limitations of ART
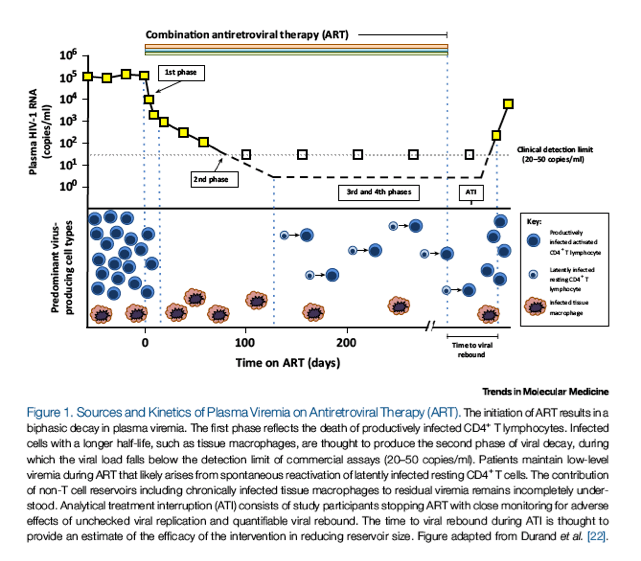
The introduction of combination ART represents a groundbreaking achievement in the effort to combat HIV-1 infection [1, 2]. Durable blockade of viral replication by combinations of antiretroviral drugs has transformed HIV-1 infection from a lethal condition characterized by progressive immune deficiency into a manageable medical problem [3, 4, 5, 6, 7]. However, long-term ART does not result in HIV-1 eradication because of the presence of long-lived viral reservoirs (see Glossary) [8, 9, 10]. ART cessation results in viral rebound within a matter of weeks that arises from resting memory CD4+ T cells harboring HIV-1 proviral DNA integrated into the cellular genome (Figure 1). This reservoir does not decay significantly during the lifespan of an HIV-1-infected patient [11, 12]. These latently infected cells are thought to sporadically reactivate, leading to derepression of silenced HIV-1 [13]. The process likely gives rise to the low-level viremia observed in patients on ART, and is thought to be the source of productive infection and viral rebound in those who stop taking antiretrovirals [14, 15]. Multiple ART intensification trials have resulted in no change in residual viremia [16, 17, 18, 19], which underscores the need for strategies that directly target or suppress the latent reservoir [20].
The description of the first (and only) durable cure of HIV-1 infection [21] has invigorated HIV-1 'cure' research and has given rise to unique eradication strategies [22, 23, 24]. The mechanism(s) of reservoir eradication in the 'Berlin patient' who underwent allogeneic stem cell transplant to treat acute myelogenous leukemia with donor cells homozygous for the C-C chemokine receptor 5 (CCR5) ∼32 mutation [21], is still a matter of debate. However, the lack of CCR5 expression, the major coreceptor required for HIV-1 cellular entry, on engrafted donor immune cells is likely to have played a significant role.
Evidence from non-human primate models suggest that CCR5-deficient cells can suppress replication of CCR5-tropic virus [25]. Indeed, gene therapy approaches have been developed that disrupt the CCR5 coding sequence in patient T cells ex vivo. Autologous, CCR5-deficient T cells can then be expanded and reinfused into patients in an attempt to reduce the frequency of target cells for viral replication. A recent clinical trial evaluated the safety of this approach in vivo; this demonstrated engraftment and persistence of these cells in the circulation and in tissues months after infusion [26].
From another standpoint, recognition of key cytokines that govern T cell activation status, trafficking and homeostasis in vivo has led to several immune-based strategies to target the HIV-1 latent reservoir. Three clinical trials studying the role of administering exogenous interferon α (IFN-α) on latent reservoir dynamics are ongoing (NCT01935089, NCT01295515, NCT02227277). One trial, adding recombinant interleukin 7 (IL-7) to intensified ART regimens has been completed, but the results are not yet published (NCT01019551). The safety and efficacy of recombinant IL-15 in reducing reservoir size will be evaluated in an approved clinical trial that is not yet open for enrollment (NCT02191098).
The focus of this review is a pharmacologic approach to reservoir elimination, also known as the 'shock and kill' strategy [27], in which ART could be supplemented for a discrete time-period with drugs that selectively reawaken dormant viruses in the latent reservoir (induced proviral transcription) and render infected cells susceptible to virus-induced apoptosis or immune-mediated clearance. After depletion of the latent reservoir, ART could then be stopped without subsequent return of viremia, resulting in a functional cure, depending on the near-complete or partial elimination of viral sequences, to be contrasted with a sterilizing cure. Several classes of latency-reversing agents (LRAs) have been intensively studied, and thorough reviews have recently been published describing the characteristics of mechanistically distinct LRA classes [23, 28, 29].
Despite considerable scientific and therapeutic advances in the three decades since the discovery of HIV-1, persistent inequities in global resource allocation and modest gains in terms of disease prevention underscore the urgent necessity of adjunctive strategies to augment ART. A pharmacologic approach to eliminate the latent reservoir with LRAs may represent a scalable strategy with the potential to turn the tide of the AIDS epidemic [30, 31]. A small number of LRAs have now reached pilot clinical trials. The rationale, design, execution, and results of these studies are reviewed here, with particular attention to the different means by which outcomes are measured, unique aspects of the trials themselves, and where the field is heading in light of the results of these pioneering studies.
Measuring HIV-1 Persistence
Pilot eradication trials have employed differing means of measuring latency reversal and reservoir perturbation in vivo [32, 33, 34]. These outcome measures constitute a spectrum connecting the molecular mechanisms of viral latency to its clinical phenotype (Table 1). Based on the consensus definition of HIV-1 latency as a state of transcriptionally silent but potentially inducible genomic integration, the most-proximal measure of proviral reactivation is quantification of intracellular unspliced HIV-1 RNA via RT-PCR. Unlike single-or multiple-spliced viral RNA species that may be present at low frequency without bona fide viral reactivation [35, 36], unspliced viral RNA transcripts are most likely to represent genomic RNA to be packaged into a nascent virion, and are therefore a necessary precursor for virion production.
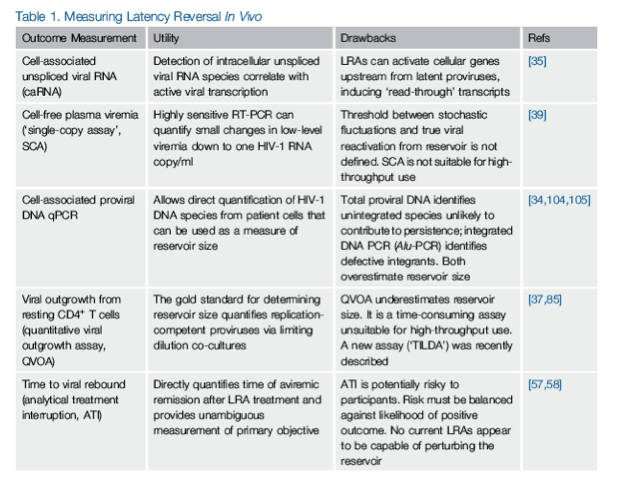
An increase in unspliced HIV-1 RNA transcripts detected by RT-PCR, often reported as a fold change over pre-intervention baseline, is a frequently employed measure for viral reactivation from latency in vivo. This measurement is not without pitfalls, however. Many LRAs, including the HDAC inhibitors, act upon host promoters upstream from transcriptionally silent proviruses, inducing transcription of cellular genes and in turn producing 'read-through' viral transcripts (Figure 2) [35, 37]. RT-PCR assays for unspliced intracellular viral RNA may be unable to distinguish these read-through transcripts, representing LRA-induced activation of cellular genes, from LRA-induced reactivation of viral transcription initiated at the authentic viral cap site. Detection of read-through transcripts decreases the specificity of this assay. For this reason, fold-change increases in unspliced intracellular HIV-1 RNA detected in vivo in the absence of other evidence for reactivation fail to rule out the possibility of an off-target LRA effect, and may not represent true latency reversal.
All patients who achieve viral suppression on ART remain viremic at levels that are below the limit of currently-available commercial viral load assays (20-50 copies of HIV-1 RNA/ml). The source of this low-level viremia, which typically fluctuates around a median frequency of one copy/ml [38, 39], is thought to be spontaneous reactivation of viral production from the latent reservoir. An increase in low-level viremia temporally related to the administration of an LRA is a reasonable surrogate for viral reactivation. A highly-sensitive RT-PCR assay, known as the single-copy assay, has been used frequently in ART intensification and eradication studies [39].
Several unanswered questions are raised by the use of the single-copy assay. Low-level viremia fluctuates in a stochastic fashion over time in patients on ART [38, 40], and highly-sensitive PCR assays to detect minute changes in viremia have the capacity to amplify these fluctuations. Similar to measurements of cell-associated viral RNA discussed above, pre-study power calculations before intervention and careful interpretation of results afterwards are necessary to reliably segregate stochastic changes in baseline low-level viremia from de novo virion release from the reactivated latent reservoir. Many studies employ both a commercial RT-PCR assay as well as the single-copy assay as a means to balance sensitivity and specificity when assaying for increases in viremia.
A detectable burst of viremia in the setting of LRA administration represents a reasonable surrogate outcome for anti-latency activity. However, it remains possible that modest transcriptional activation may occur without leading to detectable changes in viremia. Latently infected resting T cells induced to initiate proviral transcription in vivo without undergoing cellular activation may not be able to efficiently produce and release complete, infectious virions. It is likely that abundant defective proviral species will be able to reactivate and drive the expression of a subset of viral genes (those not affected by mutations) without being able to produce infectious particles [41, 42].
Importantly, release of infectious virions may not be a necessary event for these cells to be eliminated through viral cytopathic effects or immune mechanisms. An LRA that could induce viral transcription such that viral peptides are presented to effector cells could conceivably result in targeting and depletion of infected cells without virion production or a detectable change in viremia.
Measuring change in reservoir size is a short-term, proximate measure for HIV-1 eradication. While the time between ART cessation and viral rebound is variable between patients, because activation of latently infected T cells is a stochastic process [43, 44], reservoir size has been shown to correlate inversely with time to viral rebound [40, 45, 46, 47, 48, 49, 50]. A recent comparative analysis of in vivo reservoir measurement modalities identified discordance between PCR-based and viral outgrowth detection methods [34].
PCR-based methods are likely to provide an overestimate of reservoir size because most integrated proviruses are defective due to mutations or genetic deletions [41, 42]. These proviruses, although likely to be identified by sensitive PCR techniques, are unlikely to contribute to viral rebound and may not need to be a target of eradication strategies. Alternatively, virus outgrowth techniques, of which the quantitative viral outgrowth assay (QVOA) represents the gold standard, have recently been shown to underestimate the frequency of replication competent proviruses [41]. Therefore, the measurements of viral reservoir size are intimately linked to the methods used for its assessment, and this represents a key area for method development. To that end, transcriptome and proviral analysis on single cells, performed at high-throughput, may represent the next generation of technologies propelling progress in the field [51].
The consequences of the sensitivity limits of reservoir size estimation became evident in three well-publicized cases of what initially appeared to be functional cures. Two HIV-1 positive patients, who underwent allogeneic bone marrow transplant to treat lymphoma and subsequently experienced graft-versus-host disease [52], and an infant who was infected at delivery and started immediately on ART [53], experienced prolonged periods without rebound viremia after ART cessation (range 2.8-27.6 months). During these aviremic intervals, virus outgrowth from the latent reservoir could not be detected in any of these patients, raising the possibility of reservoir eradication (or lack of reservoir formation in the case of the early-ART-treated infant). However, rebound viremia ultimately developed in all three cases, indicating that HIV-1 persisted at a level sufficient to re-kindle productive infection but below the limit of detection of gold-standard reservoir assays [54, 55]. These cases were a sobering reminder of the inherent challenges of reservoir eradication.
The ultimate goal of pharmacologic HIV-1 eradication strategies is to induce prolonged remission from viral replication in the absence of antiretroviral treatment. Therefore, controlled cessation of ART, paired with close monitoring for development of viremia, represents a clinical outcome measure closely tied to the primary clinical objective with highly-desirable performance characteristics: development of viremia after treatment interruption within the time-frame delineated by multiple, well-powered treatment interruption studies is indicative of an unsuccessful anti-latency therapy. By contrast, a prolonged aviremic period off ART after administration of an LRA that extends beyond the expected time to viral rebound is highly predictive of reservoir perturbation.
The concerns raised by including analytical treatment interruption (ATI) in eradication trials arise not from interpretation of the results but rather from the potential to cause harm to participants who stop and have to re-start ART. The Strategies for Management of Antiretroviral Therapy (SMART) study, a clinical trial in which participants were randomized to continuous or intermittent ART [56], identified a significantly higher risk of opportunistic infections or death for those in the intermittent therapy arm, and has served as the basis for current Department of Health and Human Services guideline recommendations to maintain continuous and indefinite ART for all HIV-1-infected individuals. The increase in mortality with intermittent ART is likely due to highly proinflammatory effects of ongoing viral replication [56]. The decision to include ATI in an eradication trial therefore must take into account the likelihood of successful reservoir depletion by an LRA, and balance this with a trial framework that is able to identify viral rebound as early as possible, and swiftly re-initiate ART to minimize participant exposure to the proinflammatory effects of ongoing viral replication [57, 58].
First, Do No Harm: Clinical Trial Outcomes
T Cell Activation
Activated CD4+ T cells are responsible for the vast majority of HIV-1 detectable in the plasma of untreated, viremic patients, and are thought to have a lifespan of 1-2 days in vivo [59, 60], while latently infected resting memory CD4+ T cells persist for years [11, 12] and harbor proviruses that are transcriptionally silent until these cells become activated. The first clinical trials to directly target the latent reservoir were based on the recognition that the activation state of an HIV-1-infected T cell correlates directly with both the production of infectious virions and inversely with cellular lifespan [60, 61]. These trials hypothesized that activating resting T cells in vivo would lead to virion production by latently infected cells, which in turn would lead to rapid cell turnover and reservoir depletion. Participants were maintained on ART during the intervention to prevent de novo infection from occurring in bystander T cells. Four trials administering cytokines (IL-2 with or without IFN-γ) to patients on stable ART demonstrated no change in viral reservoirs or time to viral rebound when therapy was stopped [14, 62, 63, 64]. Two trials directly and more-aggressively targeted T cell activation by employing a combination of murine antibodies against human CD3 (OKT3) followed by IL-2 [65, 66].
The first OKT3 trial, by Prins et al. [66], employed multiple outcome measures including quantitation of plasma HIV-1 RNA and proviral DNA in peripheral blood mononuclear cells (PBMCs), viral outgrowth in CD4+ T cell cultures ex vivo, and direct HIV-1 RNA in situ hybridization on lymph node biopsies obtained before and after the intervention. OKT3 (5 mg) was infused daily on days 1-5, and 4.5 million IU of recombinant human IL-2 were infused twice daily on days 2-6. The trial was designed to have two sequential infusion courses 2 weeks apart.
However, two of the three participants opted out of the second course because of adverse effects. Participants experienced unremitting fever, headaches, nausea, vomiting and diarrhea throughout the six day infusion period. They also developed anemia and lymphopenia, which resolved after the infusions ceased. One participant suffered hemodialysis-dependent acute kidney injury as a result of acute tubular necrosis, as well as seizures requiring administration of anti-epileptic agents. Magnetic resonance imaging of the brain demonstrated white matter changes consistent with published reports of IL-2-induced neurotoxicity [67].
While all participants demonstrated evidence of T cell activation and development of antibodies against murine OKT3, perturbation of the latent reservoir was not observed. One participant experienced a transient increase in plasma viremia to 1500 RNA copies/ml on day 5 of infusion [67]. Of note, this participant had a low-level detectable viral load (110 RNA copies/ml) immediately before the start of the intervention. The other two participants had undetectable viral loads at enrollment and remained undetectable with respect to plasma HIV-1 RNA throughout the study. In two subjects, a mild increase in HIV-1 RNA (2-3-fold) was observed in lymph node specimens. Treatment-induced lymphopenia obscured the detection of proviral DNA levels in PBMCs and of virus outgrowth in CD4+ T cell cultures during the treatment phase, and no significant changes were observed afterward compared to baseline levels (Table 2, Key Table).
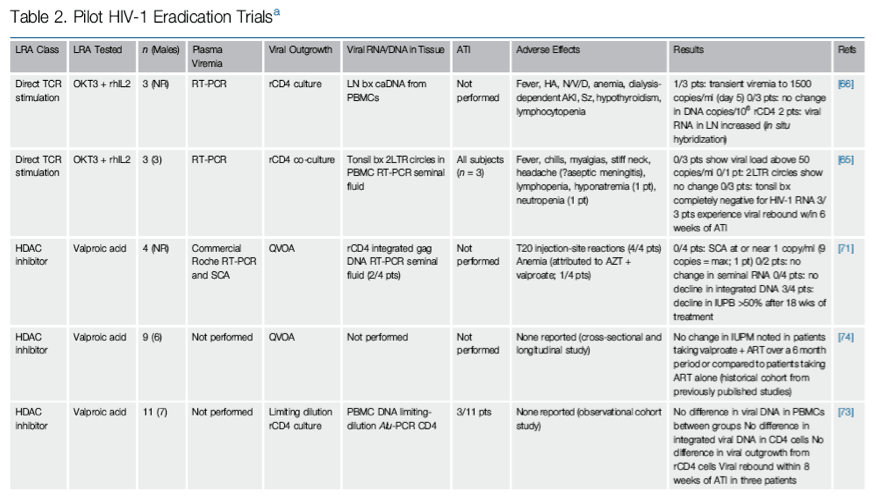
These modest results, paired with the severe adverse effects of the intervention, helped inform the design of a second pilot trial attempting to validate the anti-CD3/IL-2 strategy [65]. Kulkosky et al. administered OKT3 and IL-2 to three subjects, with notable protocol differences from Prins et al. [66]. Initially, participants underwent 'intensification' of their ART regimens with didanosine and hydroxyurea, before the study intervention, to ensure maximal viral suppression. The dosing and frequency of OKT3 were significantly reduced: instead of 5 mg daily for 5 days, Kulkosky et al. administered a 0.4 mg intravenous dose once. This was followed by administration of IL-2 (1.2 million IU/m2/day) for a 15 day period. Outcome measures were also different: for the first time, an analytical treatment interruption was included for participants in whom ex vivo viral outgrowth was below detection limits after the intervention. Plasma and seminal fluid were obtained for HIV-1 RNA quantification by RT-PCR, CD8+-depleted PBMC were obtained for co-cultures assaying viral outgrowth, and tonsil biopsies were obtained for HIV-1 RNA in situ hybridization.
Despite the lower dose and single exposure to OKT3, all three participants developed symptoms compatible with aseptic meningitis (fevers, chills, myalgias, headache, stiff neck). Headaches persisted for up to 1 week after a single infusion. Participants also developed transient lymphopenia after OKT3 (7-14 days in duration). Plasma HIV-1 RNA remained below 50 copies/ml in all subjects during the intervention. Tonsil biopsies demonstrated no evidence of viral RNA in any specimen. All three subjects agreed to participate in treatment interruption, and all developed detectable viremia over a 3-6 week period, and were re-started on ART with subsequent viral suppression.
The high toxicity and lack of efficacy observed in the OKT3 trials have informed all subsequent efforts at HIV-1 eradication using LRAs. While it is widely acknowledged today that T cell activation via T cell receptor (TCR) engagement is an untenable eradication strategy, based largely on the results of these trials, it is worth noting that these early studies built upon evidence compiled from contemporaneous pilot trials in which OKT3 and IL-2 were used for solid organ transplant and renal cell carcinoma [67, 68]. At the time of the design of these early HIV-1 eradication trials in the late 1990s, combination ART was itself a relatively novel approach to HIV-1 management that carried significant adverse effects and whose long-term efficacy was not yet assured. The availability of newer-generation ART combinations and a deeper understanding of the durability of viral suppression they provide have decreased the tolerance for potential adverse events in pilot HIV-1 eradication trials.
Valproic Acid
Studies of the molecular mechanism of viral latency have revealed the crucial role of epigenetic modifications by cellular enzymes [69]. Chromatin remodeling and, in particular, histone deacetylation by cellular histone deacetylases (HDACs) contribute to silencing of proviral gene expression [70], while inhibition of HDAC enzymes leads to increases in cell-associated viral RNA in both latency models in vitro and aviremic patient cells ex vivo [23, 28]. Preclinical evidence demonstrating that HDAC inhibitors can induce reactivation of latent proviruses without global T cell activation made these compounds attractive alternatives to OKT3 and IL-2. This drug class represents the most widely tested LRA in vivo, with four unique HDAC inhibitors having been in pilot trials to date.
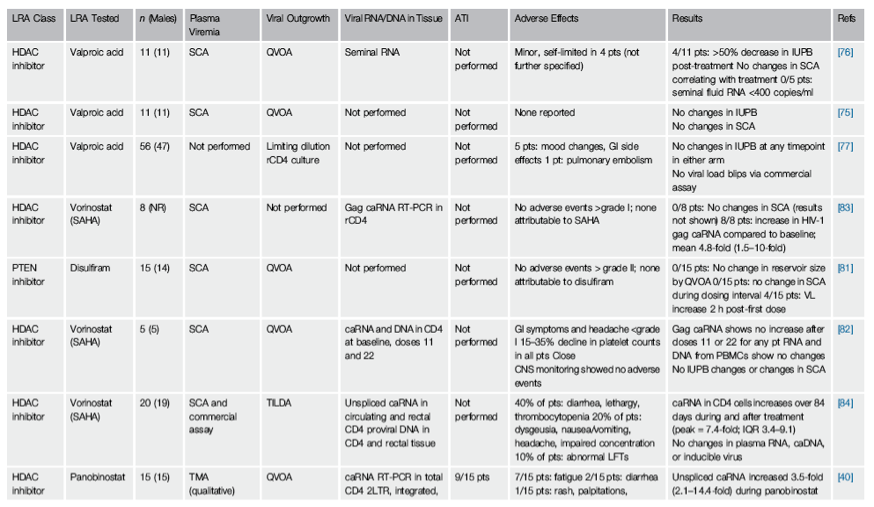
The first HDAC inhibitor to reach clinical trials was valproic acid [71]. Valproate (US brand name: Depakote) is an FDA-approved drug used to treat epilepsy and bipolar disorder. Lehrman et al. enrolled four ART-treated subjects whose regimens were intensified with the fusion inhibitor enfuvirtide (also known as T20) for 4-6 weeks before twice-daily oral administration of 500-750 mg valproate [71]. Participants were treated with valproate for 3 months, and drug dosing was individualized based on a goal plasma valproate concentration of 50-100 mg/l. Outcome measures included both commercial and single copy RT-PCR to quantify viremia, RT-PCR on seminal fluid, quantitative viral outgrowth, and integrated proviral gag DNA from circulating resting CD4+ T cells.
The trial interventions were well-tolerated by participants. Enfuvirtide, administered subcutaneously, caused injection-site reactions, and in one participant the combination of zidovudine (part of the baseline ART regimen) and valproate led to transient anemia. No changes in T cell activation profiles were observed. No significant changes occurred in low-level viremia or seminal fluid viral RNA. The frequency of resting CD4+ T cells harboring proviruses measured by viral outgrowth decreased after the 18 week intervention compared to baseline in all participants (range 29-84% decrease). The investigators concluded that HDAC inhibition appeared to lead to partial reservoir depletion in these participants. The contribution of ART intensification to this outcome was unclear and became the subject of a subsequent study by the same group (discussed below).
The Lehrman et al. study was the first published report of a targeted reduction in the HIV-1 reservoir in vivo, and generated both excitement and passionate debate in the nascent field of HIV-1 persistence research [72]. Two independent groups conducted observational studies to determine whether viral reservoirs were diminished or absent in patients who were prescribed ART as well as long-term valproate for clinical indications [73, 74]. The first of these studies, by Siliciano et al., employed a cross-sectional design in recruiting nine ART-treated individuals who had been taking valproate for a minimum of 6 months (range 6-38 months). These subjects underwent phlebotomy at two time-points (0 and 6 months) for quantification of viral outgrowth, which allowed investigators to model reservoir decay kinetics on valproate. The change in the frequency of latently infected cells in valproate-treated patients was also compared to the reservoir decay rate observed in a previously studied cohort of aviremic subjects on ART (n = 59) who were not treated with an inducing agent. These investigators did not observe any difference in the frequency of latently infected cells among patients treated with ART and valproate over time, or when this group was compared to patients on ART alone [74].
Separate work by Sagot-Lerolle et al. [73] utilized an observational study design to compare reservoir size in 11 ART-treated patients who were prescribed valproate for a median of 10 years (range 2-14 years) against 13 matched controls. Limiting-dilution viral outgrowth assays using resting CD4+ T cells were performed together with an integrated proviral DNA PCR assay. Three of the 11 valproate-treated subjects were concomitantly enrolled in ongoing structured treatment-interruption trials, allowing investigators to determine whether valproate administration influenced the kinetics of viral rebound in the absence of ART. No difference in proviral DNA from PBMCs or resting CD4+ T cells was observed, and no change in the frequency of latently infected cells quantified by viral outgrowth was identified between the two groups. All three participants who underwent ART interruption experienced detectable viremia within 8 weeks, which did not differ significantly from the median time to rebound observed in these trials.
Two prospective valproate clinical trials were subsequently conducted by the Margolis group to determine the role played by ART intensification in the setting of valproate administration [75, 76]. In the first [76], 11 subjects were recruited and 1000 mg valproate (Depakote ER) was administered daily for 16 weeks without intensifying antiretroviral agents being added to baseline ART regimens. Viral outgrowth was performed twice before and twice after the intervention. When the pooled values of each pair of reservoir measurements were compared (ART alone versus ART + valproate), only four of 11 participants experienced a decline in frequency of latently infected cells after the intervention. Single-copy assay did not demonstrate any change in low-level viremia throughout the trial. The second trial [75] included three study arms: (i) 16 weeks of valproate with baseline ART, (ii) enfuvirtide intensification and valproate, or (iii) addition of the integrase inhibitor raltegravir to baseline ART and valproate. Three participants in this 11 subject trial had participated in their previous valproate trial and received extended valproate dosing. Decreases in reservoir size observed in these subjects during the initial trial were not sustained when they were evaluated 48-96 weeks from the initial intervention. In the remaining subjects, ART intensification with enfuvirtide or raltegravir administered alongside valproate failed to have an effect on reservoir size.
Perhaps the final word on valproate came from a trial conducted by Routy et al. in which 56 patients on stable ART received 16 or 32 weeks of valproate (500 mg twice daily) [77]. No changes in latently infected cell frequency were observed relative to baseline values or between the two arms. The combined results of these prospective and observational studies provide ample evidence that valproate alone was not sufficient to perturb the latent reservoir [78]. These trials did however provide an important framework for the design of future pilot eradication studies and offered a first-hand opportunity to consider the pros and cons of outcome measures that should be employed as more efficacious HDAC inhibitors were identified.
Disulfiram
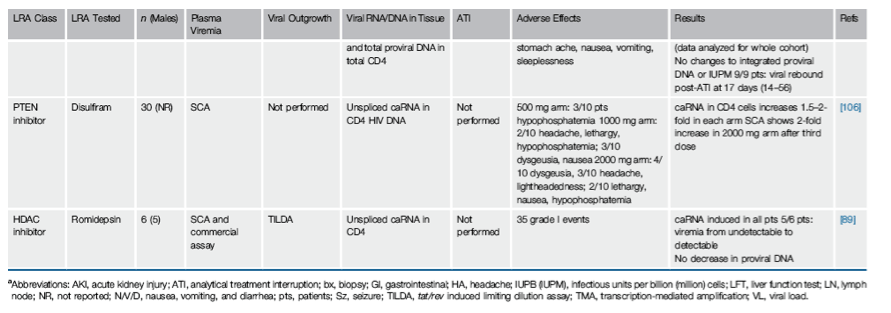
The potential for latency reversal induced by disulfiram was identified using a primary cell model of latency [79]. Disulfiram was FDA-approved for use as a deterrent to alcohol ingestion in the 1950s, and therefore has decades of clinical safety data supporting its use for novel indications in vivo. The mechanism of action with regard to latency reversal appears to be through depletion of the intracellular protein PTEN, which in turn activates the Akt signaling pathway to initiate proviral transcription in an NF-κB-dependent manner [80].
Two pilot clinical trials have tested the hypothesis that disulfiram can perturb the latent reservoir in vivo. Spivak et al. recruited 16 participants who underwent directly observed administration of 500 mg disulfiram for 14 days after baseline measurement of latent reservoir size by QVOA [81]. Single-copy assay PCR was performed every other day during the intervention and weekly afterward. A second reservoir measurement was performed 10 weeks after disulfiram was discontinued. Disulfiram concentrations were highly variable among participants, with a majority demonstrating no detectable plasma levels at any time during the administration period. Disulfiram was well tolerated without any adverse effects, however, no change in reservoir size was observed. While there was also no clear signal for an increase in low-level viremia, a minority of participants experienced viral blips within hours of disulfiram administration. It is unclear whether metabolism of disulfiram and initiation of viral transcription could lead to a systemic increase in viral load in the timeframe in which these blips were observed. A second disulfiram trial has been recently published that revealed similar results: early, transient increases in viral transcription [106]. Reservoir size was not measured. The results to date suggest that disulfiram alone is unlikely to perturb the latent reservoir.
Vorinostat
The initial results and tolerability of valproate were followed closely by the description of 'next-generation' HDAC inhibitors that have since reached clinical trials. Vorinostat, an FDA-approved chemotherapy agent also known as SAHA, has been tested in three separate trials [82, 83, 84]. The first of these [83] enrolled eight participants to undergo a single infusion of vorinostat. Careful measurement of histone acetylation confirmed the bioactivity of the drug in vivo, and RT-PCR demonstrated a mean 4.8-fold increase in cell-associated HIV-1 RNA 6 h after vorinostat dosing (range 1.5-10-fold). No changes in low-level viremia or reservoir size were observed. In a follow-up study involving some of the same participants, Archin et al. [82] evaluated the effect of 22 doses of vorinostat on intracellular viral RNA levels. A positive signal observed after a single dose in the previous study [83] was not identified in this study [82]. Cell-associated viral RNA showed no changes after doses 11 or 22 in the five participants enrolled. No changes in low-level viremia, proviral DNA, or reservoir size were observed.
In a separate trial evaluating multiple doses of vorinostat, Elliott et al. [84] demonstrated similar results as the initial single-dose study [83]. Twenty participants were administered 400 mg vorinostat daily for 2 weeks. Adverse events were common in this study: 40% of participants experienced diarrhea, lethargy, and thrombocytopenia, 20% developed nausea, vomiting, dysgeusia (alteration of taste sensation), and headache, and 10% had changes in liver function testing. No changes were observed in plasma viremia, cell-associated proviral DNA, or reservoir size measured by a novel PCR-based method, the tat/rev induced limiting dilution assay or TILDA [85]. Cell-associated viral RNA increased 7.4-fold (mean value, inter-quartile range 3.4-9.1-fold), and in many participants (9/20) the peak of intracellular RNA production was observed after the dosing interval. This trial made use of high-throughput sequencing techniques to evaluate the transcriptional changes induced during vorinostat administration and those changes that persisted afterward. Interestingly, upregulation of genes associated with protein ubiquitination and major histocompatibility complex (MHC) class I antigen presentation was identified 10 weeks after vorinostat administration. The lack of change in viremia and reservoir size led these authors to conclude that vorinostat alone is unlikely to purge the latent reservoir.
Panobinostat and Romidepsin
Panobinostat is an FDA-approved chemotherapy agent used in the treatment of multiple myeloma [86, 87]. A pilot eradication clinical trial in Denmark by Rasmussen et al. enrolled 15 patients to receive 20 mg of panobinostat three times per week during weeks 1, 3, 5, and 7 of an 8 week study [40]. This intermittent dosing schedule allowed investigators to evaluate pharmacodynamics with regard to histone acetylation and cell-associated unspliced viral RNA. Adverse effects were minor, consisting mostly of fatigue (7/15 participants) and diarrhea (2/15). Histone acetylation increased during each dosing interval and returned to baseline during the washout periods, while cell-associated viral RNA increased rapidly after the first dose (mean 2.4-fold increase, range 1.8-3.3) and appeared to increase during each of the dosing intervals. Contemporaneous plasma viremia was measured using a qualitative transcription-mediated amplification assay, and the investigators observed an increase in the percentage of participants with assays positive for plasma HIV-1 RNA during panobinostat administration compared to baseline (54% versus 30%). Total and integrated HIV-1 DNA in CD4+ T cells showed no change during the trial, and QVOA showed no change in the frequency of latently infected cells after panobinostat administration compared to baseline. Despite the lack of significant changes in reservoir size, nine participants elected to undergo ART interruption after panobinostat administration. All participants became viremic with a median time to viral rebound of 17 days (range 14-56 days), which is within the range of viral rebound observed in multiple treatment interruption trials.

Romidepsin is an HDAC inhibitor FDA-approved for the treatment of T cell lymphoma that has shown to be more potent in reversing latency in vitro than vorinostat or panobinostat [88]. In a recent trial by S�gaard et al. [89], romidepsin was administered to six patients at a dose of 5 mg/m2 intravenously once per week for 3 weeks. Thirty-four grade I adverse events and two grade II events (fatigue and fever) were described. Cell-associated RNA was induced in all participants during the treatment period, and in five of six participants low-level viremia became detectable during the trial period. No decrease in proviral DNA was observed. Further in vivo studies will be required to finalize the verdict on romidepsin.
Future Directions
Evidence of low-level viral transcription paired with lack of reservoir perturbation (measured by viral outgrowth, proviral PCR quantitation, or viral rebound after ART discontinuation) are all findings common to these early HIV-1 eradication trials. What accounts for the modest in vivo efficacy of LRAs tested to date? A variety of primary T cell latency models, in which uninfected healthy donor T cells are infected in vitro with HIV-1 and progress to a latently infected state, have been developed independently and used to identify potential LRAs. These results in turn have informed the design of various pilot HIV-1 eradication trials described here. A recent comparative analysis of LRA activity across these published in vitro latency models identified significant inter-model discordance [90]. The results of this study indicate that no current in vitro latency model faithfully recapitulates the nature of the HIV-1 latent reservoir. Preclinical research on LRA efficacy and toxicity gathered from non-human primate SIV models [91, 92], humanized mouse models [93, 94, 95, 96], and purified resting CD4+ T cells obtained from HIV-1 infected, aviremic patients [97, 98] are current strategies addressing this significant knowledge gap (Box 1, Box 2).
Pharmacologic strategies for HIV-1 eradication have sought to minimize T cell activation largely in response to the unacceptable toxicity of the OKT3 and cytokine administration trials [65, 66]. HDAC inhibitors have been an attractive choice for pilot eradication trials because they offer an acceptable balance between proviral transcriptional activation and cellular activation. However, the modest outcomes of these clinical trials have led to a re-evaluation of these agents as lead compounds. In two separate ex vivo analyses, LRAs from the protein kinase C (PKC) family, bryostatin-1 [35] and ingenol 3,20 dibenzoate [98], demonstrated significantly higher latency-reversal potential than the HDAC inhibitors tested. Bryostatin-1 also demonstrated synergy with HDAC inhibitors when used together ex vivo [97]. The accumulation of recent ex vivo results are helping to make a case for PKC agonists, potentially including bryostatin-1 or ingenol derivatives, as candidates for pilot eradication trials. Combination LRA trials making use of multiple anti-latency mechanisms, particularly HDAC inhibition and PKC activation, should also be a priority. Several groups have begun to explore the potential for synergy when LRAs from unique mechanistic classes are combined [97, 99, 100]. In these in vitro studies, PKC agonists appear to hold promise when combined with other agents. Further research is required to understand the potential for off-target effects of these combinations, including the induction of proinflammatory cytokine secretion.
PKC agonists act upon T cell activation pathways and upregulate cell surface markers including CD69. The effect of T cell activation induced by administration of PKC agonists in vivo cannot be predicted from these experiments, although Laird et al. demonstrated that, despite upregulation of activation markers, these cells did not produce significant levels of inflammatory cytokines [97]. Of note, in the ex vivo analysis that preceded the clinical trial of romidepsin, CD69 expression increased significantly in resting CD4+ T cells exposed to clinical concentrations of this drug, a finding unique to romidepsin among HDAC inhibitors [88]. Perhaps the efficacy observed with romidepsin in vitro and in vivo reflects an adjunctive mechanism beyond HDAC inhibition that involves T cell activation.
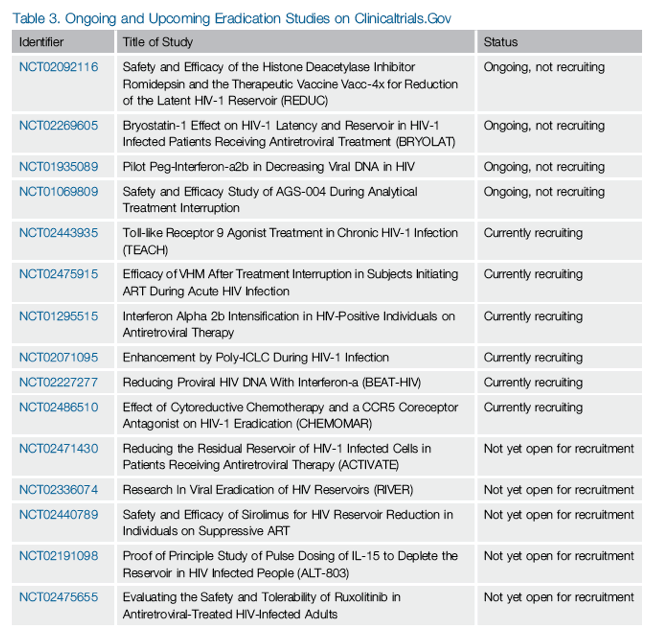
Ongoing trials listed at clinicaltrials.gov at the time of this review include several studies revisiting the use of cytokines as well as Toll-like receptor agonists to perturb the reservoir, a second romidepsin trial, and the first in vivo study of bryostatin-1 as an LRA (Table 3). Novel gene-therapy techniques including the use of CRISPR/Cas9 technology are showing great potential [101], and will hopefully reach clinical trials in the near future. A unique cure strategy has been recently proposed by Mousseau et al. [102, 103] making use of an inhibitor of the HIV-1 accessory protein Tat to suppress proviral transcription in latently infected cells. The mechanistic diversity of these approaches holds promise for the future of HIV-1 eradication research.
Concluding Remarks
While pilot eradication trials have not achieved the ultimate goal of a durable ART-free virologic remission, a promising trajectory can be observed from early interventions characterized by high toxicity and lack of efficacy to current studies making use of well-tolerated short course therapies that appear to induce proviral transcription. These encouraging early signals offer potential therapeutic building-blocks and can serve to highlight the larger unanswered questions in the field of HIV-1 eradication.
In the face of the challenges presented by viral latency, measurable and incremental progress has been made in these early eradication studies. Large questions remain with regard to the ideal agent or combination of agents necessary to perturb the reservoir, as well as to what are the most reliable means of quantifying latency reversal and reservoir depletion (see Outstanding Questions). The collaborative efforts of research groups around the globe and the support of funding agencies committing resources to this crucial health problem allow an optimistic outlook despite the complexity of HIV-1 persistence and the modest gains achieved to date. 'It is not the end', remarked Winston Churchill in his famous 1942 address, balancing cautious optimism with the recognition of a lengthy and costly struggle to come. 'It is not even the beginning of the end. But it is, perhaps, the end of the beginning.'
Outstanding Questions
What are the most predictive measurements of reservoir perturbation in vivo? PCR-based methods to detect proviral transcription can be overly sensitive and lead to difficulty in distinguishing between stochastic fluctuations and minor LRA-induced reservoir perturbations. However, the current gold-standard for quantifying reservoir size, quantitative viral outgrowth, lacks the sensitivity to detect small amounts of dormant proviruses in vivo.
Is analytical treatment interruption (ATI) appropriate for HIV-1 eradication pilot trials? If not, what are acceptable surrogate measures of reservoir depletion? ATI represents the most direct measure of the likelihood of a functional cure. However, this exposes participants to the known risks of unchecked viral replication.
Are there adjunctive strategies to attenuate the potential toxicity of LRAs that induce T cell activation in vivo? Early HIV-1 eradication trials demonstrated that activating T cells in vivo led to significant adverse events. However the LRAs that consistently demonstrate the highest efficacy across HIV latency models lead to T cell activation.
Will adjunctive strategies be necessary for clearance of cells harboring proviruses reactivated by LRAs? None of the LRAs that have reached clinical trials have demonstrated perturbation of the latent reservoir. Reservoir depletion is likely to be a prerequisite for functional cure using the 'shock and kill' pharmacologic approach.
Acknowledgments
The authors gratefully acknowledge critical input from colleagues Laura Martins and Alberto Bosque in the preparation of this manuscript. This review was supported in part by ongoing funding from the National Center for Advancing Translational Sciences of the National Institutes of Health under award number 1KL2TR001065 (A.M.S.). This work is dedicated to the memory of Dr Frederick L. Brancati, an inspirational and incomparable mentor.
|
|
| |
| |
|
|
|