| |
Comparing Generic and Innovator Drugs: A Review of 12 Years of Bioequivalence Data from the United States Food and Drug Administration
|
| |
| |
Download the PDF here
Abstract
BACKGROUND In the US, manufacturers seeking approval to market a generic drug product must submit data demonstrating that the generic formulation provides the same rate and extent of absorption as (ie, is bioequivalent to) the innovator drug product. Thus, most orally administered generic drug products in the US are approved based on results of one or more clinical bioequivalence studies.
OBJECTIVE To evaluate how well the bioequivalence measures of generic drugs approved in the US over a 12-year period compare with those of their corresponding innovator counterparts.
METHODS This retrospective analysis compared the generic and innovator bioequivalence measures from 2070 single-dose clinical bioequivalence studies of orally administered generic drug products approved by the Food and Drug Administration (FDA) from 1996 to 2007 (12 y). Bioequivalence measures evaluated were drug peak plasma concentration (Cmax) and area under the plasma drug concentration versus time curve (AUC), representing drug rate and extent of absorption, respectively. The generic/innovator Cmax and AUC geometric mean ratios (GMRs) were determined from each of the bioequivalence studies, which used from 12 to 170 subjects. The GMRs from the 2070 studies were averaged. In addition, the distribution of differences between generic means and innovator means was determined for both Cmax and AUC.
RESULTS The mean ± SD of the GMRs from the 2070 studies was 1.00 ± 0.06 for Cmax and 1.00 ± 0.04 for AUC. The average difference in Cmax and AUC between generic and innovator products was 4.35% and 3.56%, respectively. In addition, in nearly 98% of the bioequivalence studies conducted during this period, the generic product AUC differed from that of the innovator product by less than 10%.
CONCLUSIONS The criteria used to evaluate generic drug bioequivalence studies support the FDA's objective of approving generic drug formulations that are therapeutically equivalent to their innovator counterparts.
Generic pharmaceutical products play a vital role in US healthcare. Since the passage of the Drug Price Competition and Patent Term Restoration Act in 1984 (Hatch-Waxman Amendments),1 which set the rules under which generic drugs could compete with innovator products, the Food and Drug Administration (FDA) has approved 11,843 generic drug products. As of December 2008, a total of 13,239 prescription and over-the-counter drugs were listed as marketed in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations ("Orange Book"),2 of which 8893 (67.2%) were listed as "multisource" products (meaning that generic versions are marketed). Generic drugs offer a powerful approach to cost-savings for the patient-in 2008, generic drugs accounted for 69% of all prescriptions dispensed in the US, yet only 16% of all dollars spent on prescriptions.3
All prescription and over-the-counter generic drugs marketed in the US must meet standards established by the FDA. In approving a new generic drug for marketing, the FDA concludes that it is therapeutically equivalent to its corresponding reference product (usually the innovator product, but sometimes another generic product if the innovator product was withdrawn). The FDA believes that therapeutically equivalent drug products can be substituted with the full expectation that both products will produce the same clinical response.2 A generic drug is approved by the FDA if it is:
1. pharmaceutically equivalent to an approved safe and effective reference product in that it (a) contains identical amounts of the same active drug ingredient in the same dosage form and route of administration and (b) meets compendial or other applicable standards of strength, quality, purity, and identity;
2. gbioequivalent to the reference product in that it (a) does not present a known or potential bioequivalence problem and it meets an acceptable in vitro standard (usually dissolution testing) or (b) if it does present such a known or potential problem, it is shown to meet an appropriate bioequivalence standard;
3. adequately labeled; and
4. manufactured in compliance with current Good Manufacturing Practice regulations.2
The regulatory oversight of generic drug chemistry, manufacturing, and controls is identical to that imposed upon innovator drug products.4
Despite the wide use of generic drugs in US health care and the stringent regulatory standards governing generic drug approval for marketing, generic substitution continues to be a topic of heated debate among healthcare professionals, members of the pharmaceutical industry, consumers, and government officials.5-10 For example, there are concerns about small numbers of reported cases of breakthrough seizures or an increased seizure frequency in patients who switch from brand-name to generic antiepileptic drugs.11 Likewise, there are concerns about the findings of a retrospective analysis which showed that, when 975 Israeli patients were switched from Coumadin (warfarin sodium) to a generic warfarin sodium clathrate, higher doses of the generic formulation were needed to maintain previously stabilized international normalized ratio (INR) values in some patients.12 The American Academy of Neurology opposes generic substitution of anticonvulsant drugs for the treatment of epilepsy without the attending physician's approval.13 Similarly, a report by the American Society of Transplantation recommended that generic immunosuppressive medications should be clearly labeled and distinguishable from innovator drugs and that patients should be educated to inform their physicians of any switch to or among generic alternatives.14
In particular, controversy continues to surround the FDA's methods for assessing in vivo bioequivalence between the generic and innovator products.5 Some scientists and clinicians have expressed the opinion that the FDA's current bioequivalence standards may not be sufficient for certain patient populations being treated with certain classes of drugs (notably, antiepileptic drugs and/or drugs with a narrow therapeutic index), drugs that display variable absorption patterns, or drugs with nonlinear pharmacokinetics.15-17
Some of the controversy arises from misunderstanding of the FDA's statistical methods for determining bioequivalence.5-7,18 This article discusses the rationale underlying the FDA's present approach for determining in vivo bioequivalence and presents data from bioequivalence studies of generic drugs approved over a 12-year period to evaluate how the FDA's bioequivalence approach performs in ensuring that the pharmacokinetic profiles of generic drugs closely approximate those of the innovator products.
The FDA requires an applicant submitting an Abbreviated New Drug Application (ANDA) for a generic drug to demonstrate that its product is bioequivalent to the corresponding reference "listed drug."1 Bioequivalence means that the generic drug and the reference drug will reach the systemic circulation at an equivalent relative rate and extent-in other words, the 2 drug products' dosage forms should produce equivalent drug concentration-time profiles in the blood. Generic applicants submitting ANDAs for systemically active solid oral dosage forms are required to submit one or more bioequivalence studies in which human subjects are given the generic or reference product and drug concentrations in the blood are measured and analyzed statistically. The ANDA process as established by the Hatch-Waxman Amendments does not require generic drug manufacturers to submit nonclinical or clinical studies to establish the safety and efficacy of the active ingredient. This is because these safety and efficacy data were previously documented during the approval process for the innovator product. It is assumed that, if the active ingredient was shown to be safe and effective after it is absorbed into the bloodstream, any drug product giving rise to blood concentrations of active ingredient to the same rate and extent will produce the same effect.
Bioequivalence studies are generally conducted in healthy male and female adults under standardized conditions. In some cases it is necessary to use patients for reasons of safety (eg, bioequivalence studies of oncology drug products are conducted in patients with cancer).19 Most bioequivalence studies use a 2-way crossover design. If the drug has a long plasma half-life, it may be more suitable to use a randomized parallel study design.20,21
The appropriate number of subjects for a bioequivalence study can be determined based on previous knowledge of the innovator drug's pharmacokinetic variability. In general, the number of subjects should be adequate to detect a 20% difference in the measured bioequivalence parameters with 80% certainty.22 The FDA recommends that investigators enroll a minimum of 12 subjects23-most studies submitted to the Office of Generic Drugs enroll from 24 to 36 subjects. The FDA asks investigators to conduct single-dose bioequivalence studies because it has been shown that these are more sensitive to detecting differences in formulation performance than are multiple-dose studies.20,21,24-27 However, it is sometimes necessary to conduct a multiple-dose bioequivalence study at steady-state, for example, if the bioequivalence study is conducted in patients.28
It is recognized that a drug's maximum concentration (Cmax) is an indirect measure of the rate of absorption; for example, changes in the rate of absorption influence Cmax only minimally.5 In addition, Cmax is influenced both by the blood sampling scheme and by the extent of absorption, and as a result there is a strong correlation between total area under the plasma concentration versus time curve (AUC) and Cmax. Cmax tends to be more variable than AUC in bioequivalence studies.29 Nonetheless, the FDA considers Cmax to be the most clinically appropriate parameter for assessing the rate of absorption and may relate to a drug's toxicity and/or efficacy.20,30
Most bioequivalence studies are conducted on the highest strength of a drug product line, unless it is necessary to use a lower strength for safety reasons. Use of the highest strength is particularly critical for drugs that display nonlinear kinetics because of nonlinear (usually capacity-limited) elimination or presystemic metabolism, for which the extent of absorption increases more than proportionally with an increase in dose.31,32 For such drugs, small differences in the rate of absorption can have substantial effects on the AUC. Thus, using the highest strength, or, in some cases, the highest starting dose-so that pharmacokinetics are in the nonlinear range-in bioequivalence studies33 ensures that a generic formulation will not pass bioequivalence acceptance criteria unless it is formulated to provide nearly the same rate and extent of exposure as the corresponding reference product. For drugs for which rate and/or extent of absorption increases less than proportionally with an increase in dose,32,34 the bioequivalence study will be most discriminating if conducted at the lowest strength or, if only one strength is marketed, at the lowest recommended dose.
Two products are deemed bioequivalent if the 90% confidence intervals of the geometric mean generic/innovator (or test/reference) Cmax and AUC ratios fall within the bioequivalence limits of 80-125%.23 To obtain geometric means, the data are log-transformed prior to conducting an analysis of variance (ANOVA), then back-transformed before calculating the test/reference ratios.
The FDA asks investigators to log-transform Cmax and AUC for bioequivalence analysis for 2 reasons. First, the ANOVA used to conduct the bioequivalence statistics is based on a linear statistical model. However, the form of expression for AUC suggests a multiplicative model, since AUC = (F*D)/(V*Ke), where F is the fraction of drug absorbed, D the dose, V the volume of distribution, and Ke the elimination rate constant.35,36 For this reason, FDA statisticians concluded that effects on AUC are not additive if the data are analyzed on the original scale of measurement. Thus, since ln(AUC) is equal to ln(F) + ln(D) - ln(V) - ln(Ke), logarithmic transformation of AUC allows it to be analyzed using the ANOVA, which assumes a linear statistical model. A similar argument can be made for Cmax.
The second reason for log transformation is that Cmax and AUC, like much biological data, correspond more closely to a log-normal distribution than to a normal distribution.35 Plasma concentration data and derived pharmacokinetic parameters tend to be skewed, and their variances tend to increase with the means. Log transformation generally remedies this situation and makes the variances independent of the means. In addition, skewed frequency distributions are often made more symmetrical by log transformation.
It is often incorrectly stated that because the width of the bioequivalence limits is 80-125%, the FDA allows Cmax and AUC to vary by -20 to +25% between products. In fact, such a large absolute difference between test and reference bioequivalence parameters will virtually always result in failure to meet bioequivalence limits. The statistical approach used by the FDA to analyze bioequivalence study data is designed to minimize the risk in situations where the patient is switched to a generic version of a medication that he or she is currently taking.
The 2 one-sided tests procedure is used to analyze bioequivalence parameter data.37 One test verifies that the bioavailability of the generic product is not more than 20% less than that of the innovator product. The other test verifies that the bioavailability of the innovator product is not more than 20% less than that of the generic product. The use of the 20% criteria is based on a decision by FDA medical experts that, for most drugs, a ±20% difference in the concentration of active ingredient in the blood will not be clinically significant.2 Numerically, this is expressed as a limit of 80% on the test mean/reference mean for the first statistical test and a limit of 80% on the reference mean/test mean for the second statistical test. Since by convention, all data are expressed as the test/reference ratios for Cmax and AUC, the limit expressed in the second statistical test becomes the reciprocal of 80%, which is 125%. As a result, the bioequivalence limits are 80-125%. Both Cmax and AUC must meet the bioequivalence limits. The determination of bioequivalence using this approach is termed average bioequivalence.37
Use of the average bioequivalence method ensures that the rate and extent of drug absorption from a generic product will differ very little from that of its innovator counterpart. If the true average response of the generic product in the population is close to 20% below or 25% above the innovator average response, one or both of the 90% confidence interval limits is likely to fall outside of the bioequivalence limits, and the product will fail the bioequivalence test (Figure 1). In fact, it has been suggested that, using the 2 one-sided tests procedure, when the mean Cmax and AUC responses of 2 drug products differ by more than 12-13%, they are unlikely to meet the bioequivalence limits of 80-125%.22
The current practice is to carry out the 2 one-sided tests at the 0.05 level of significance. Since a 5% statistical error is allowed at both the upper and lower bioequivalence limits, the combined total error is 10%, generating the 90% confidence interval of 80-125%. The practice of carrying out the 2 one-sided tests at the 0.05 level of significance ensures that, if the average bioequivalence parameters of the generic product are truly 20% less or 25% greater than corresponding innovator values, the generic will have less than a 5% chance of being approved as equivalent.18
It is logical to ask why bioequivalence data are not analyzed by a 2-tailed test at the conventional level of p less than 0.05, as opposed to performing 2 one-sided tests. In fact, in the early years of the FDA's generic drug review program, bioequivalence statistics were conducted using 2-tailed hypothesis testing at the 5% level of significance. The null hypothesis was that the means of the 2 formulations did not differ significantly. It was found that this approach resulted in a problem with the power of the test.36-39 Products that showed nearly the same means, with very small variance, could show a significant difference and be rejected. Alternatively, products that showed large differences with a large variance could show a nonsignificant difference and be deemed equivalent. To overcome these problems, the FDA began to use a power approach, which added the additional requirement that the power of the test for no difference had to be sufficiently large (80%). However, there were additional problems with the power approach. For example, if the within-subject variability was high, it would not be possible to conclude equivalence no matter what the difference between test and reference products was, because in this situation, the estimated power was less than 80%.37,39
The problems with these approaches arose from the fact that the t-test of the hypothesis of no difference does not assess the evidence in favor of the conclusion that the test and reference means are equivalent, but rather assesses the evidence in favor of a conclusion that the test and reference means are different, which is not the question of interest in the bioequivalence analysis.37,39 The 2 one-sided tests procedure currently used by the FDA resolves the problems of hypothesis testing and assumes that test and reference products within 20% of each other with respect to Cmax and AUC are bioequivalent.36
The FDA does not ask ANDA applicants to use statistical procedures to compare test and reference time to Cmax (tmax) values. Although theoretically a relatively sensitive measure of absorption rate, tmax is thought to have shortcomings as an indirect measure of the rate of drug absorption.40,41 For example, ANOVA analysis cannot be applied to tmax; unlike Cmax and AUC, which are continuous variables, tmax is a discrete measure dependent on frequency of blood sampling.42 In addition, most pharmacokinetic studies typically employ irregular sampling schemes to collect tmax data; as a result, these data are not routinely amenable to proper statistical evaluation.43 For these reasons, the FDA has decided not to impose bioequivalence acceptance criteria on the parameter tmax.20 Nonetheless, the FDA believes that tmax should be considered in bioequivalence decision-making and routinely examines tmax data in bioequivalence studies as supportive data to verify that the test and reference products have the same rate of absorption.44
Methods
COMPARING GENERIC AND INNOVATOR PRODUCT BIOEQUIVALENCE PARAMETERS
We hypothesized that, over a long period of time, generic drug product average bioequivalence parameters will differ very little from those of their corresponding innovator counterparts because of the rigors imposed by the average bioequivalence statistical approach and bioequivalence limits of 80-125%. To test this hypothesis, we collected a set of bioequivalence parameter data from all acceptable single-dose bioequivalence studies of generic solid oral dosage form drug products approved from 1996 through 2007 (12 y). This time period was selected because the Office of Generic Drugs began archiving ANDA reviews electronically in 1996, thus facilitating data collection (prior to 1996, paper copies of all ANDA reviews were stored in file rooms). Studies enrolled both male and female subjects; in all studies, the test or reference product was administered following an overnight fast. All bioequivalence studies were acceptable in that the bioequivalence parameters met the FDA's bioequivalence limits.
BIOEQUIVALENCE DATA ANALYSIS
The original pharmacokinetic and statistical calculations for the in vivo bioequivalence studies were performed by scientific reviewers from the Division of Bioequivalence, Office of Generic Drugs, using SAS (SAS Institute, Inc., Cary, NC). For the purpose of this retrospective study, these data were collected using Access (Microsoft Corp., Redmond, WA). Data analyses were conducted in Excel (Microsoft Corp., Redmond, WA) and S-Plus (TIBCO Software, Inc., Palo Alto, CA).
BIOEQUIVALENCE PARAMETERS EVALUATED
Bioequivalence parameter data included the test/reference geometric mean ratios (point estimates) for Cmax, AUC0-t, and AUC∞ from each of these studies in our dataset. The parameter AUC0-t is the area under the drug plasma concentration versus time curve from time 0 (immediately after dosing) until the last study sampling time (t); AUC∞ is AUC0-t extrapolated to infinity. The FDA Office of Generic Drugs deems a bioequivalence study acceptable only when the 90% confidence intervals of the test/reference geometric mean ratios for all 3 parameters meet the 80-125% bioequivalence limits. All bioequivalence studies evaluated for this investigation were acceptable to the Office of Generic Drugs in that the 90% confidence intervals of the test/reference geometric mean ratios for all 3 parameters met bioequivalence limits. Until recently, generic drug applicants were not required to submit unacceptable bioequivalence studies to ANDAs. As of July 15, 2009, ANDA applicants are required to submit data from all bioequivalence studies on a drug product formulation submitted for approval.45
To determine how well each generic drug (test) product performed compared with its corresponding innovator (reference) product, we performed 2 types of comparisons.
COMPARING GENERIC/INNOVATOR GEOMETRIC MEAN RATIOS
For the first comparison, we first obtained the geometric mean test/reference ratio for Cmax, AUC0-t, and AUC∞ from each bioequivalence study in our data set. The parameter Cmax is used here to illustrate how the geometric mean ratios are calculated. First, the individual test (TEST) and reference (REF) Cmax values are ln-transformed. The averages are calculated for lnCmaxTEST and lnCmaxREF. The geometric mean ratios are then determined by taking the antilogarithm of the difference between the average lnCmaxTEST and average lnCmaxREF. In practice, in the Office of Generic Drugs, the geometric mean ratios are calculated using ANOVA, general linear models procedure (PROC GLM) available in SAS.46 The ESTIMATE statement in SAS PROC GLM is used to obtain estimates for the adjusted differences between test and reference treatment means. The antilogarithm of the ESTIMATE gives the test/reference ratio in the normal scale.
Thus, the equation for calculating a geometric mean ratio for the parameter Cmax in a bioequivalence study is as follows:
where
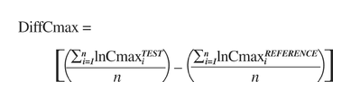
The same procedure is used to calculate geometric mean ratios for AUC0-t and AUC∞.
For the first comparison, we determined the Cmax, AUC0-t, and AUC∞ geometric mean ratios from each of the acceptable bioequivalence studies and averaged these geometric mean ratios.
COMPARING ABSOLUTE DIFFERENCES BETWEEN GENERIC AND INNOVATOR GEOMETRIC MEANS
For the second comparison, we determined the absolute differences between the test and reference Cmax, AUC0-t, and AUC∞ geometric means. The geometric mean is the antilogarithm of the average of the ln-transformed Cmax, AUC0-t, and AUC∞ values for the test and reference products. We compared the magnitude of these differences to see how close the generic drug product geometric means were to the innovator drug product geometric means.
COMPARING IMMEDIATE-RELEASE AND MODIFIED-RELEASE PRODUCTS
We compared data from bioequivalence studies of immediate-release (IR) and modified-release (MR) solid oral dosage forms. The term modified-release encompasses extended-release (also referred to as sustained- or controlled-release) and delayed-release products.
Results
We collected data from a total of 2070 acceptable single-dose bioequivalence studies of solid oral dosage forms, conducted from 1996 to 2007. The number of subjects used in these studies ranged from 12 to 170. The geometric mean (test/reference) ratios for 2070 bioequivalence studies of solid oral dosage form generic drugs approved during that time averaged 1.00 for all 3 bioequivalence parameters (Table 1). The average percent differences between the test and reference geometric means for Cmax, AUC0-t, and AUC∞ were 4.57 ± 3.59%, 3.23 ± 2.74%, and 3.17 ± 2.69%, respectively (Table 2).
We determined the average geometric mean (generic/innovator) ratios and mean differences between generic and innovator bioequivalence measures for IR and MR products to compare performance of the 2 types of oral dosage forms. Our data set consisted of 1788 (86%) and 282 (14%) bioequivalence studies conducted for IR and MR products, respectively. Geometric mean ratios in the bioequivalence studies of IR products averaged 1.00 for Cmax, AUC0-t, and AUC∞, respectively (Table 1). Results were similar for the bioequivalence studies of MR products; the average of all geometric mean ratios was 0.99 for Cmax, and 1.00 for AUC0-t and AUC∞, respectively. For MR products, the mean difference between the generic and innovator geometric mean ratios was slightly greater than for IR products (Table 2).
Figure 2 shows a histogram depicting the distribution of geometric mean test/reference ratios for Cmax, AUC0-t, and AUC∞ for the 2070 bioequivalence studies. The generic drug differed from its corresponding reference drug by less than 10% in 91.5% and 97.6% of the studies for Cmax and AUC0-t, respectively (Table 3). In only 49 (2.4%) of the 2070 bioequivalence studies evaluated, the generic and innovator AUC0-t values differed by more than 10%. In only 3 (0.15%) of the 2070 studies, generic and innovator AUC0-t values differed by 15-16%.
In general, differences between generic and innovator bioequivalence parameters were somewhat lower for IR products than for MR products (Figure 3). For Cmax, the generic and innovator differed by less than 10% in 92.3% and 87.5% of the studies for IR and MR products, respectively (Table 3). For AUC0-t, the generic and innovator differed by less than 10% in 97.8% and 96.1% of the studies for IR and MR products, respectively (Table 3).
We recognized that the tables and figures, which represent the distribution of mean ratios and differences, do not consider sample size variability among the studies. For example, the larger the sample size, the better the estimate of mean difference or ratio. Since there is a relationship between sample size and the accuracy of the estimate of the mean difference or ratio, the within-subject variability associated with each drug product is an important variable that should be taken into account. Different drug products can vary significantly in the degree of variability, confounding the relationship between sample size and the accuracy of the estimate. To address this concern, we created scatter plots of the percent difference between means and sample size for each of the parameters. Percent difference was used to normalize for different plasma concentration units used in the different studies, as well as different bioavailabilities of the drug substances. The resulting scatter plots did not appear to show a relationship between sample size and the difference between means.
Additionally, distribution plots of effect sizes for the parameters were created. Effect size was calculated as the difference between geometric mean test and reference values divided by the standard deviation of the reference value, (meantest - meanreference)/SDreference. The distribution of effect sizes did not suggest any directional bias. This is consistent with the fact that the bioequivalence studies evaluated for this paper are for different drugs and different formulations and are conducted by different investigators. As such, it would be highly unlikely that test products would consistently show greater or less bioavailability than reference products.
Discussion
Our findings confirm the results of 2 similar FDA reviews comparing bioequivalence measures. A review of 224 in vivo bioequivalence studies in ANDAs approved shortly after the Hatch-Waxman amendments were passed, from 1984 to 1986, found that the average percent difference between mean AUCs of the innovator drug and generic drug was about 3.5%.47 A review of 127 in vivo bioequivalence studies of generic drugs approved in 1997 found mean percent differences between the innovator and generic products of 4.29%, 3.47%, and 3.25% for Cmax, AUC0-t, and AUC∞, respectively.48
We evaluated plausible reasons why, in 2.4% of the studies, the generic AUC0-t varied from the reference AUC0-t by more than 10%. For this evaluation, we considered AUC0-t to be the most meaningful of the 3 bioequivalence parameters. It is well established that AUC is less variable than Cmax,29 which depends upon the sampling schedule and varies (particularly for IR products) with the magnitude of AUC.5 Of the 2 total exposure parameters, AUC0-t is considered a more accurate representation of systemic exposure than AUC∞, as AUC0-t is determined by experimental measurement, whereas a portion of AUC∞ is derived by mathematical extrapolation.49
The 49 studies for which the generic AUC0-t differed from the reference by more than 10% were of 39 different drugs from a variety of drug classes. None is considered a narrow therapeutic index drug; in addition, none of these was an antiepileptic or immunosuppressant drug. We surveyed the formulations of these products to determine whether the test and reference products contained differing amounts of excipients known to affect bioavailability, such as alcohol sugars,50 polysorbate-80,51 or β-cyclodextrins.52 We were unable to identify any properties of the excipients used that could result in differences in bioavailability between the test and reference products.
However, most of these 39 drugs were previously identified as highly variable drugs, that is, as having high within-subject variability (≥30%) in the bioequivalence measures AUC and Cmax. In general, these drugs are characterized as having poor aqueous solubility, having low oral bioavailability, and/or undergoing extensive first-pass metabolism. A description of these highly variable drug substances, including mechanism of action, physicochemical properties, and pharmacokinetic characteristics, has been published.53 It is difficult for highly variable drugs and drug products to meet the standard bioequivalence criteria using a reasonable number of study subjects. Thus, for generic highly variable drugs, the FDA presently recommends that ANDA applicants increase the number of study subjects, use a group-sequential study design, or use a reference-scaled average bioequivalence approach. In the reference-scaled approach, the bioequivalence criterion is scaled to the within-subject variability of the reference product in a crossover bioequivalence study, together with a constraint imposed on the geometric mean ratio between the test and reference products. The reference-scaled approach is supported by the pharmaceutical science community because it is believed that highly variable drugs generally have a wide therapeutic window; in other words, despite high variability, these products have been demonstrated to be both safe and effective.54
Our retrospective analysis has the advantage that it encompassed results of a large number of acceptable bioequivalence studies (2070) submitted to the Agency in support of generic drug products approved over a long time period (12 y). Nonetheless, information is lacking about some factors that could influence the variability of the data collected. First, it is not possible to compare the variability of the data from acceptable bioequivalence studies in our study with the variability of data from unacceptable bioequivalence studies conducted during the same time period. This is because, until recently, ANDA applicants were not required to submit all bioequivalence studies conducted on the final to-be-marketed formulation. The Agency did not begin to receive results from an appreciable number of unacceptable bioequivalence studies until after a Pharmaceutical Sciences Advisory Committee meeting in 2000, at which ANDA applicants were urged to submit the results of all bioequivalence studies on the final generic drug formulations.55 Thus, although the Office of Generic Drugs has received some data from unacceptable bioequivalence studies, we presently have no way of determining whether these data are from all unacceptable bioequivalence studies or represent a biased sample. Since ANDA applicants are now required to submit data from all bioequivalence studies on a drug product formulation submitted for approval,45 it will be possible for the FDA to conduct comparative analyses of acceptable and unacceptable bioequivalence study data.
A second factor possibly influencing the variability of the data in our study is that, for many of the studies evaluated, the majority of subjects were healthy individuals between the ages of 18 and 60. Until 2000, when the FDA first posted its Guidance for Industry on Bioavailability and Bioequivalence Studies for Orally Administered Drug Products, bioequivalence studies on generic drug products generally enrolled exclusively healthy young, male subjects. FDA Guidance for Industry now recommends that investigators enroll in bioequivalence studies individuals representative of the general population, taking into account age, sex, and race, with an emphasis on (1) recruiting similar proportions of males and females in the study and (2) including as many subjects of 60 years or older as possible.20 Thus, many investigators now enroll in bioequivalence studies similar proportions of males and females and subjects from various ethnicities. However, very few studies include subjects aged 60 years or older. In addition, bioequivalence studies conducted at clinical sites in some Asian countries continue to enroll only males of one race. It is not known what effect further increasing the diversity of the study population would have on the distribution of variability in Cmax and AUC ratios.
One factor that could support the validity of the FDA bioequivalence approach would be an analysis of postmarketing safety and efficacy data on approved generic drug products. Future research on the performance of the FDA's generic drug program could involve an evaluation of MedWatch generic drug data submitted to the FDA. It should be emphasized that the FDA has access to all safety data obtained during the bioequivalence study. Scientific reviewers from the Office of Generic Drugs evaluate the adverse event data from the test and reference products. If a reviewer notices a difference in the incidence of adverse events during the study, the ANDA is forwarded to an FDA medical officer for a clinical consult. Bioequivalence studies are deemed unacceptable if there is a clinically significant difference between test and reference products with respect to adverse events. Thus, for all the studies compared in this retrospective analysis, the nature and severity of adverse events experienced by the subjects were comparable in the test and reference product treatment groups.
Our evaluation of 12 years of acceptable bioequivalence studies suggests that, overall, pharmacokinetic measures of drug rate and extent of exposure differ very little between innovator drugs and corresponding generic drugs approved for marketing in the US. Consequently, the FDA expects that switching to a generic drug from the innovator drug will not affect clinical outcome. Findings from several clinical studies of switches from innovator to corresponding generic products support this hypothesis. In a prospective, observational study, a health maintenance organization that formerly dispensed only Coumadin monitored 182 enrollees for 8 months prior to and 10 months after a switch to a generic warfarin sodium tablet product and found no significant changes in INR, frequency of INR monitoring, number of dose changes, and rate of thrombic and hemorrhagic events.56 In an observer-blinded, crossover study, no significant difference in INR or adverse event profiles was observed in patients who had received Coumadin for at least 2 months and were randomized to receive 28-day periods of generic warfarin sodium tablets for 1 period followed by Coumadin for 2 periods or Coumadin for 1 period followed by generic warfarin sodium tablets for 2 periods.57 A recent study of hospital admission rates for cardiovascular diseases among 49,673 users of brand-name and generic metoprolol did not reveal any differences in the incidence rates of serious cardiovascular events between the brand-name and generic group after confounder adjustment.58 A recent aggregate meta-analysis of 47 studies compared 8 subclasses of generic and innovator cardiovascular drugs; the analysis concluded that the generic and innovator drugs were similar in nearly all clinical outcomes, including vital signs, clinical laboratory values, adverse events, and healthcare system utilization.59 Clinical equivalence was noted in 100% of β-blockers, 91% of diuretics, 71% of calcium channel blockers, 100% of antiplatelet agents, 100% of statins, 100% of angiotensin-converting enzyme inhibitors, and 100% of α-blockers. In this same study, among narrow therapeutic index drugs, clinical equivalence was reported in 100% of class 1 antiarrhythmic agents and 100% of warfarin.
Nonetheless, substitution of generic antiepileptic, immunosuppressant, and narrow therapeutic index drugs continues to be a controversial subject. For a number of reasons, the FDA's position is that less-expensive generics can be substituted for the innovator (more-expensive) drug products, regardless of the indication. First, as shown by this study and previous retrospective studies, the rate and extent of drug absorption differ very little between innovator drugs and corresponding generic drugs approved for marketing in the US. Thus, the FDA believes that, considering the variability of the overall clinical response, any contribution of generic substitution to varying plasma concentrations is negligible. For example, high variability in antiepileptic drug plasma concentrations has been reported in patients on continuous treatment.60,61 In addition, coadministration of certain drug products without dose adjustment (as recommended according to product labeling) and lack of patient adherence may cause changes in drug plasma concentrations exceeding any small changes that might result from generic substitution.16
Another important reason why the FDA believes that approved generic drugs may be substituted for their corresponding innovator products is that ANDA applicants are required to submit the same drug substance and drug product CMC (Chemistry, Manufacturing, and Controls) information for approval as NDA applicants.62Both NDA and ANDA applicants must submit to the FDA a full description of the drug substance, including its physical and chemical characteristics and stability; the method of synthesis (or isolation) and purification of the drug substance; the process controls used during manufacture and packaging; and specifications needed to ensure the identity, strength, quality, and purity of the drug substance. Both NDA and ANDA applicants must submit to the FDA a list of all components used in the manufacture of the drug product (regardless of whether they appear in the drug product); specifications for each component to ensure its quality; product design and development, manufacturing, and process control; and specifications needed to ensure the identity, strength, quality, purity (including impurities), and bioavailability of the drug product. The FDA does not approve NDA and ANDA products if the Agency determines that the submitted CMC information is not acceptable.
Another reason that the FDA believes that generics can be safely substituted for the corresponding innovator products is that, throughout the lifetime of a generic product, the FDA will carefully investigate any reports of therapeutic inequivalence and take regulatory action if necessary. Two recent cases illustrate this process. In the first case, due to reports of clinical concerns about the quality of various levothyroxine sodium tablets (which many consider to be a narrow therapeutic index drug), the FDA initiated actions to narrow the potency specifications for all levothyroxine sodium tablet products.63 The United States Pharmacopeia subsequently endorsed the new narrow potency specifications for this product.64 These narrow potency specifications, now required by the FDA, ensure that both generic and innovator levothyroxine sodium tablets will maintain potency, and consequently clinical effect, throughout the approved shelf life.
In the second case, due to claims that a generic version of cyclosporine was not bioequivalent to the corresponding innovator product under some conditions of use, the FDA promulgated a new bioequivalence regulation. A firm that manufactured a generic cyclosporine oral solution had conducted a study showing that its product was not bioequivalent to the corresponding innovator product when both were administered in apple juice (the products were bioequivalent when administered in water) but did not inform the FDA of this study finding until 2 years after approval.65 To prevent this situation from happening again, the FDA immediately took steps to promulgate a new rule requiring ANDA applicants to submit all bioequivalence studies on the final to-be-marketed formulation, whether the studies meet or fail to meet bioequivalence acceptance criteria. The new rule, Requirements for Submission of Bioequivalence Data, was issued in final form in 2009.45 The new rule amends the Code of Federal Regulations (CFR) Bioavailability/Bioequivalence Regulations of 21 CFR Part 320 to require an ANDA applicant to submit data from all bioequivalence studies that an applicant conducts on a drug product formulation submitted for approval, including studies that do not meet the specified bioequivalence criteria. The final rule also amends portions of 21 CFR Part 314 (Applications for FDA Approval to Market a New Drug) Subpart C, Abbreviated Applications. All bioequivalence studies submitted on the same drug formulation as that submitted for approval must be submitted to the FDA either as a complete study report or a summary report of the bioequivalence data. The term same drug product formulation means that the formulation of the drug product submitted for approval and any formulations that have minor differences in composition or method of manufacture from the formulation submitted for approval are similar enough to be relevant to the Agency's determination of bioequivalence. Thus, this new rule will provide additional assurance that generic products are therapeutically equivalent to their corresponding innovator counterparts by allowing the FDA to make regulatory decisions based on all bioequivalence data obtained for a given product.
The above examples show that the FDA takes very seriously claims of therapeutic inequivalence of generic drug products, including antiepileptic, narrow therapeutic index, and immunosuppressant drugs, and will take necessary steps to investigate such claims and modify regulatory requirements. The FDA continues to regulate generic drug products even after the initial approval process to ensure therapeutic equivalence to the corresponding innovator products.
A final consideration supporting generic substitution is the fact that generic drugs are generally considerably less costly than the corresponding innovator products. The use of generic drug products has resulted in savings of $734 billion to the US healthcare system over the past decade, with $121 billion of these savings achieved in 2008 alone.66 One study estimates that the enactment of legislation preventing a pharmacist from substituting generic for innovator counterparts of drug classes such as antiepileptics, immunosuppressants, and antipsychotics would result in increased drug costs to commercial payors by $17.5 billion, Medicaid by $6.2 billion, and consumers by $5.3 billion over 10 years.67
As the new administration of the US government seeks to cut health costs, an attractive approach is to initiate a regulatory pathway for approving generic versions of biologic drug products. Notably, several countries have set a precedent for such approvals.68 The European Union (EU) opened up a regulatory pathway for "biosimilars" in 2005, and since then its European Medicines Agency has approved 13 such drugs. A number of the European guidelines have been adopted in Australia, and Japan this year issued its own guideline for the regulation of biosimilars. The World Health Organization and Health Canada have recently issued draft guidelines that generally follow the EU model. In the US, however, the pathway for approval of "follow-on" biologics is stalled, in part because the law that governs licensing of biologic products does not contain a provision for follow-on biologics. The issue is currently before the US Congress. Discussion in Congress so far focuses on a number of key issues that must be addressed in any approval system for follow-on biologics, including duration of market exclusivity for the innovator product, scope, data requirements, immunogenicity interchangeability, trade names, and economic considerations.69 One important issue under debate is how much discretion the FDA should have to determine requirements for approval of follow- on biologics.
To summarize, the FDA's generic drug policies are designed to approve high-quality generic drug products that are therapeutically equivalent to their innovator counterparts. Both generic and innovator products must meet the same FDA standards for manufacturing and quality. Despite concerns about the rigor of FDA's bioequivalence testing methods, the FDA's record shows that its bioequivalence approach works quite well to ensure that drug plasma concentrations achieved after dosing with generic drug products differ very little from drug plasma concentrations observed after dosing with the corresponding innovator counterparts. In our survey of generic drugs approved over a 12-year period, the average difference in the rate and extent of drug absorption between generic and innovator products was 4.35% and 3.56%, respectively. In addition, in nearly 98% of the bioequivalence studies conducted during this period, the extent of drug absorption from the generic product differed from that of the innovator product by less than 10%. These findings emphasize that the bioequivalence statistical criteria used for generic drug approvals effectively preclude large differences between products that the FDA deems as bioequivalent.
The robust performance of bioequivalence testing in generic drug approvals over many years lends strong support to the FDA's belief that health professionals can substitute drug products determined to be therapeutically equivalent with the full expectation that the generic product will produce the same clinical effect and safety profile as the innovator product.
|
|
| |
| |
|
|
|