 |
|
|
| |
Elafibranor - (GFT505) NASH Treatment - GOLDEN 505 Study & Commentary
|
| |
| |
Download the PDF here
Download the PDF here
This report contains the published study and the Commentary....
Elafibranor, (GFT505), an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening
"In conclusion, this randomized controlled trial provides evidence that pharmacologic modulation of the PPARα/δ nuclear receptors results in substantial histologic improvement in NASH, including resolution of steatohepatitis and improvement of the cardiometabolic risk profile, with a favorable safety profile. Larger phase 3 trials of elafibranor in the target population of patients with moderate to severe NASH are warranted."
--------------------
Entering the GOLDEN Age for Therapies in NASH - Commentary/Editorial
PHILIP N. NEWSOME
Institute for Health Research (NIHR)
Birmingham Liver Biomedical Research Unit and Centre for
Liver Research
University of Birmingham and
Liver Unit
University Hospital Birmingham NHS Foundation Trust
Birmingham, United Kingdom

The burden from nonalcoholic fatty liver disease (NAFLD) is increasing rapidly,1 as reflected in referrals to hospital specialists as well as consideration for liver transplantation,2 and yet there are no licensed therapies for this condition. In this edition of Gastroenterology, Ratziu et al3 report on the efficacy of GFT505, a dual peroxisome proliferator-activated receptor (PPAR)-α and -δ agonist, in a well-conducted phase II study in patients with nonalcoholic steatohepatitis (NASH). GFT505 has been shown previously to have potent activity in preclinical models of NAFLD, reducing hepatic lipid accumulation as well as decreasing expression of proinflammatory and profibrotic genes.4 The PPAR-α receptor is highly expressed in rodent hepatocytes where, after agonism, it prevents accumulation of triglycerides through an induction of genes involved in mitochondrial and peroxisomal fatty acid β-oxidation.5However, GFT505 was still able to decrease steatosis and liver inflammation in a PPARα knockout mouse fed a Western diet and with a more pronounced antifibrotic effect.4 The latter suggests that PPAR-δ agonism may mediate much of the antifibrotic effect of GFT505, which is supported by the high levels of PPAR-δ expression in hepatic stellate cells, which are known to increase further during hepatic stellate cell activation and liver fibrosis.6 The effects of PPAR-δ agonism on steatosis and inflammation can also be explained by its ability to stimulate hepatic fatty acid β-oxidation6 and its induction of macrophages to adopt an alternative antiinflammatory M2 phenotype.7 Early phase clinical studies supported the efficacy of GFT505 as demonstrated by an improvement in liver biochemistry when administered to patients with type 2 diabetes. These studies also provided important insights on the mechanism of action of GFT505, demonstrating improvements in peripheral and hepatic insulin sensitivity with associated decreases in plasma free fatty acid levels.8
To assess more directly efficacy in NASH, Ratziu et al randomized 274 noncirrhotic patients with biopsy-proven NASH to either oral elafibranor (GFT505) 80 or 120 mg daily or to placebo for 52 weeks. The study did not meet its predefined primary endpoint, which was reduction of any of the components of the NAFLD Activity Score (NAS)9 to zero without worsening in fibrosis (progression to stage 3 or 4). However, post hoc analyses of patients with higher baseline NAS (≥4) did demonstrate superiority of the 120 mg elafibranor dose versus placebo (20% vs 11%; P = .018). Also, in response to changing recommendations for the assessment of efficacy in NASH trials, the investigators included a further post hoc analysis after completion of the study to assess the number of patients clearing NASH as defined by disappearance of ballooning together with either disappearance of lobular inflammation or the persistence of mild lobular inflammation (score of 0 or 1). As before, this had to be without worsening in liver fibrosis, although this was now defined as progression by ≥1 stage. In this latter analysis, a greater proportion of patients receiving 120 mg elafibranor met this secondary post hoc endpoint compared with those on the placebo (19% vs 12%; P = .045), with the difference being more pronounced for patients with a higher baseline NAS (≥4; 19% vs 9%; P = .013). In keeping with previous phase II studies, more than one-half of the patients in this study (51%) had F0/F1 fibrosis, raising questions about the response rate in patients with more advanced liver fibrosis, which are now becoming the focus of phase III registration trials. Notably, the efficacy of elafibranor 120 mg in patients with NAS ≥ 4 and F1-3 fibrosis (22% resolution vs 13%; P = .026) was also seen in patients with NAS ≥ 4 and F2/3 (15% resolution vs 9% in placebo; P = .002), although in keeping with an earlier study, which suggested lower absolute response in those with advanced fibrosis.10
Primary endpoints in NASH have evolved over the past few years and have been helped by consensus guidelines such that there is now acceptance that resolution of NASH without any worsening in liver fibrosis is a standard phase 2b endpoint.11 The definition of resolution of NASH can take several forms ranging from a primarily NAS-based assessment to a more overarching gestalt assessment of the presence, or not, of NASH. Thus, despite the laudable convergence toward an accepted phase II primary endpoint, there remain challenges with the precise definition as it remains an inherently subjective assessment.12 Another strength of this study is the central reading of slides at inclusion and at the end of the study, which is necessary to decrease the impact of this subjective component on study outcome. Intuitively, an assessment by >1 pathologist would further increase confidence of any observed outcome although there are challenges in either colocating expert pathologists or transferring slides across the globe. In that regard, the use of digital photographs of sections, if and when approved by regulatory authorities, would ease some of these logistical hurdles, allowing simultaneous evaluation and discussion.
Notwithstanding the caveats around assessing histological primary endpoints in NASH, it is helpful to review the collective experience to ascertain the current reported levels of efficacy. As indicated in Table 1, a wide range of primary endpoints have been utilized in the past few years rendering it difficult to make direct comparisons of efficacy across treatments. Moreover, although many of the studies have included resolution of NASH as a secondary endpoint, no studies have included the concomitant need for no worsening in liver fibrosis. Thus, rates of NASH resolution for pioglitazone (47%),13 vitamin E (36%),13 and obeticholic acid (22%),14 where no stipulation on change in fibrosis was made, are not directly comparable with those reported for liraglutide (39%)10 or elafibranor (19%),3 where fibrosis change was also taken into account. Ultimately, it is the progression to advanced fibrosis/cirrhosis that determines the development of clinical complications of liver disease, and it remains unclear what level of impact on hepatic inflammation/NASH is required to prevent this.
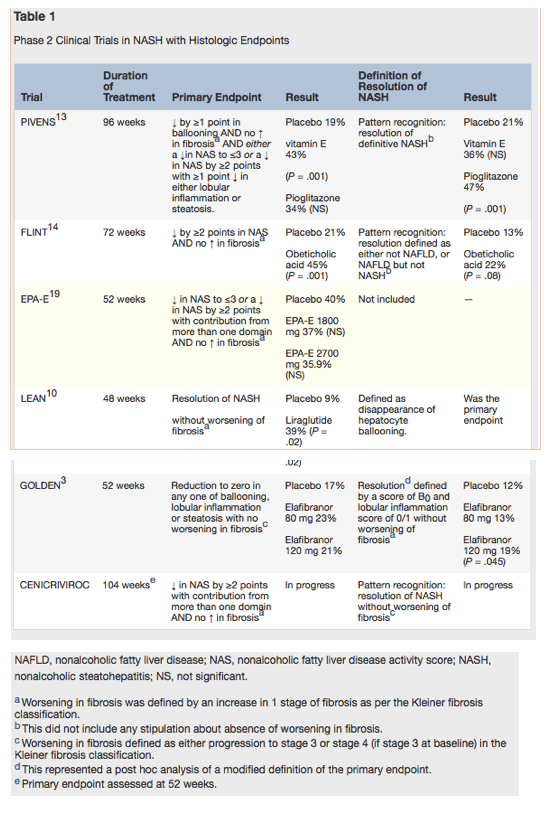
Notably, the focus on NASH reflects the perception that it epitomizes the injurious process, which is at the heart of progressive disease in patients with NAFLD, and yet recent studies have presented data suggesting that clinical outcomes are predicted by the level of fibrosis rather than the presence of NASH.15, 16 It is, of course, possible that these studies are potentially confounded by their retrospective nature, or that they fail to capture the dynamic nature of the presence/absence of NASH histologically, although progress in science does require consideration of challenges to accepted dogma. This uncertainty is perhaps reflected in consensus documents with input from regulatory authorities that require hard clinical endpoints or nonprogression to cirrhosis for full approval of agents in NASH.11
Given the significant cardiovascular morbidity and mortality found in patients with NAFLD,15 it is important to consider the impact of any therapeutic agent on these outcomes. Elafibranor improved lipid parameters (high- and low-density lipoprotein cholesterol and triglycerides), glycemic control as well as systemic inflammatory markers such as highly sensitive C-reactive protein. Notably, there were no cardiovascular events or deaths with elafibranor, which is particularly encouraging as other studies, with similar age patients, have suggested cardiovascular event rates on placebo ranging from 12 in 8313 and 9 in 142.14 The decrease in renal function in 7 patients treated with elafibranor is a potential concern given the known association of chronic kidney disease with NASH17 and this will need to be monitored in longer term studies.
In conclusion, this study adds another therapeutic dimension for patients with NASH, and we await the outcome of pivotal phase III studies to determine its ultimate role. It is an exciting time for therapies in NASH, and although success in many other conditions has come from combinatorial therapies, most studies in NASH have examined just a single agent. As synergistic effects cannot necessarily be predicted from monotherapy studies, perhaps now is the time to take heed of previous advice18 and make the leap and undertake combination studies.
---------------------------
Elafibranor, (GFT505), an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening
Gastroenterology May 2016 - Vlad Ratziu,1,2 Stephen A. Harrison,3 Sven Francque,4 Pierre Bedossa,5 Philippe Lehert,6,7 Lawrence Serfaty,8 Manuel Romero-Gomez,9 Jérôme Boursier,10 Manal Abdelmalek,11 Steve Caldwell,12 Joost Drenth,13 Quentin M. Anstee,14 Dean Hum,15 Remy Hanf,15 Alice Roudot,15 Sophie Megnien,15 Bart Staels,16 and Arun Sanyal,17 on behalf of the
GOLDEN-505 Investigator Study Group
Background & Aims
Elafibranor is an agonist of the peroxisome proliferator-activated receptor-α and peroxisome proliferator-activated receptor-δ. Elafibranor improves insulin sensitivity, glucose homeostasis, and lipid metabolism and reduces inflammation. We assessed the safety and efficacy of elafibranor in an international, randomized, double-blind placebo-controlled trial of patients with nonalcoholic steatohepatitis (NASH).
Methods
Patients with NASH without cirrhosis were randomly assigned to groups given elafibranor 80 mg (n = 93), elafibranor 120 mg (n = 91), or placebo (n = 92) each day for 52 weeks at sites in Europe and the United States. Clinical and laboratory evaluations were performed every 2 months during this 1-year period. Liver biopsies were then collected and patients were assessed 3 months later. The primary outcome was resolution of NASH without fibrosis worsening, using protocol-defined and modified definitions. Data from the groups given the different doses of elafibranor were compared with those from the placebo group using step-down logistic regression, adjusting for baseline nonalcoholic fatty liver disease activity score.
Results
In intention-to-treat analysis, there was no significant difference between the elafibranor and placebo groups in the protocol-defined primary outcome. However, NASH resolved without fibrosis worsening in a higher proportion of patients in the 120-mg elafibranor group vs the placebo group (19% vs 12%; odds ratio = 2.31; 95% confidence interval: 1.02-5.24; P = .045), based on a post-hoc analysis for the modified definition. In post-hoc analyses of patients with nonalcoholic fatty liver disease activity score ≥4 (n = 234), elafibranor 120 mg resolved NASH in larger proportions of patients than placebo based on the protocol definition (20% vs 11%; odds ratio = 3.16; 95% confidence interval: 1.22-8.13; P = .018) and the modified definitions (19% vs 9%; odds ratio = 3.52; 95% confidence interval: 1.32-9.40; P = .013). Patients with NASH resolution after receiving elafibranor 120 mg had reduced liver fibrosis stages compared with those without NASH resolution (mean reduction of 0.65 ± 0.61 in responders for the primary outcome vs an increase of 0.10 ± 0.98 in nonresponders; P < .001). Liver enzymes, lipids, glucose profiles, and markers of systemic inflammation were significantly reduced in the elafibranor 120-mg group vs the placebo group. Elafibranor was well tolerated and did not cause weight gain or cardiac events, but did produce a mild, reversible increase in serum creatinine (effect size vs placebo: increase of 4.31 ± 1.19 μmol/L; P < .001).
Conclusions
A post-hoc analysis of data from trial of patients with NASH showed that elafibranor (120 mg/d for 1 year) resolved NASH without fibrosis worsening, based on a modified definition, in the intention-to-treat analysis and in patients with moderate or severe NASH. However, the predefined end point was not met in the intention to treat population. Elafibranor was well tolerated and improved patients' cardiometabolic risk profile.
Of all chronic liver diseases, nonalcoholic steatohepatitis (NASH) is of increasing concern, as it is highly prevalent, potentially severe and without approved therapy. NASH defines a subgroup of nonalcoholic fatty liver disease where liver steatosis coexists with hepatic cell injury (apoptosis and hepatocyte ballooning), and inflammation.1 It occurs in close association with overweight/obesity, type 2 diabetes, and cardiometabolic conditions that define the metabolic syndrome.2 Because of the prevalence of these comorbidities, NASH is emerging as the most common chronic liver disease.
NASH promotes liver fibrosis and some patients progress to severe hepatic diseases, including cirrhosis, liver failure, hepatocellular carcinoma, or require liver transplantation.3, 4 Liver-related mortality is increased 10-fold in NASH patients compared with the general population.5 However, NASH is also a multisystem disease that could worsen insulin resistance, the metabolic syndrome, and the systemic inflammatory state.6 Consequently, NASH patients also have an increased rate of cardiovascular events and neoplasia. These 2 latter conditions carry the heaviest toll in terms of mortality, the leading cause of death being from cardiovascular events.3, 7, 8
Peroxisome proliferator-activated receptors (PPARs) are nuclear receptors playing key roles in cellular processes regulating metabolic homeostasis, immune-inflammation, and differentiation. PPARγ agonists demonstrated efficacy in improving histology in NASH,9, 10, 11 but side effects, such as congestive heart failure, peripheral edema, bone fractures, and weight gain severely restrict their prescription and acceptance as long-term therapies. PPARα is most prominently expressed in the liver and is activated by hypolipidemic fibrates. PPARα controls the lipid flux in the liver by modulating fatty acid transport and β-oxidation, while improving plasma lipids by decreasing triglycerides and increasing high-density lipoprotein (HDL) cholesterol.12 In addition, PPARα activation inhibits inflammatory genes induced by nuclear factor-κB and decreases the expression of acute-phase response genes.12PPARδ (also called PPARβ) regulates metabolism in liver and peripheral tissues. PPARδ agonists enhance fatty acid transport and oxidation, increase HDL levels, and improve glucose homeostasis by enhancing insulin sensitivity and inhibiting hepatic glucose output.13 Importantly, PPARδ exerts anti-inflammatory activities in macrophages and Kupffer cells.14 In a pilot trial, a selective PPARδ agonist reduced liver fat content, while improving insulin sensitivity, plasma lipids, and decreasing γ-glutamyltransferase.15
Elafibranor (GFT505) is a dual PPARα/δ agonist that has demonstrated efficacy in disease models of nonalcoholic fatty liver disease (NAFLD)/NASH and liver fibrosis.16 Elafibranor confers liver protection by acting on several pathways involved in NASH pathogenesis, reducing steatosis, inflammation, and fibrosis. In phase 2a trials in dyslipidemic, prediabetic and type 2 diabetic patients, elafibranor consistently improved plasma lipids and glucose homeostasis, peripheral and hepatic insulin resistance, and reduced liver inflammatory markers.17, 18
This phase II study was conducted to assess the efficacy of elafibranor for NASH in an international, randomized, placebo-controlled, multicenter, 1-year clinical trial.
Discussion
This randomized controlled trial provides evidence of efficacy of the dual PPARα/δ activator elafibranor on both histologic reversal of NASH and metabolic improvement in patients with NASH. Both are important objectives on the path of controlling NASH. Steatohepatitis is indirectly associated with reduced hepatic survival in NAFLD.5, 23 It drives fibrogenesis, a slow process of hepatic scar formation that can result in cirrhosis and its deadly complications, such as liver failure, portal hypertension, and hepatocellular carcinoma. Consequently, clearance of steatohepatitis,24 that is, reversal to a normal liver or to steatosis without steatohepatitis-a condition not associated with increased hepatic morbidity or mortality-is expected to improve hepatic prognosis and is now accepted as the best, short-term surrogate for histologic improvement in NASH trials.25, 26
When analyzed according to the a priori, protocol-defined primary outcome, there were no significant differences in treatment response between the 2 elafibranor groups and placebo. However, when using an updated, modified definition of reversal of NASH without worsening of fibrosis,19, 20 the 120-mg elafibranor arm performed significantly better than placebo in the ITT population. The latter definition is more stringent than the one used in the protocol. First, it emphasizes hepatocyte ballooning, a sign of liver-cell injury and a cardinal feature of steatohepatitis that is associated with disease progression and enhanced fibrogenesis. In contrast, the protocol-based definition required the disappearance of either steatosis, inflammation, or hepatocyte ballooning. Second, based on older data showing that bridging fibrosis but not earlier stages is associated with liver-related mortality, 23, 27, 28, 29 only progression to bridging fibrosis (or to cirrhosis) was considered "worsening of fibrosis" in the protocol-based definition. Instead, the modified definition defines worsening of fibrosis as any one-stage increase based on recent data showing that even early fibrosis is associated with global and liver-related mortality.7 Importantly, this more stringent definition led to a lower placebo effect. Earlier studies have not explicitly defined reversal of NASH and subtle differences in the criteria used might explain the variable rates of response in the placebo group (from 13%21 to 21%11). Therapeutics in NASH is an evolving field and previous trials have used an aggregate histologic score, the NAS, as a primary end point.11, 21 However, the prognostic value of the NAS is not established.7, 23, 30 We expect that future, large phase 3 trials will be using this more stringent definition of response and, therefore, we reported on both definitions of primary response in an attempt to facilitate comparisons of the magnitude of the effect both across trials and across classes of pharmacologic agents. Interestingly, for both definitions there was a significant interaction effect with baseline activity, suggesting that the latter is an important determinant of the efficacy of elafibranor.
Regardless of the definition of response, elafibranor at 120 mg was significantly superior to placebo in the post-hoc analysis after excluding the 15% of patients with mild steatohepatitis (ie, bNAS of 3). The 80-mg dose was not significantly better than placebo in any primary or secondary histologic analyses. Patients with mild but well-defined NASH were allowed to participate because of early concerns about recruitment feasibility, and because it was assumed that resolution of NASH was dependent on the presence of NASH and not on a particular level of severity. In these patients with mild steatohepatitis, there was an unexpectedly high placebo response rate that might have led to a lack of treatment effect in the planned primary outcome assessment. In addition, the observation that elafibranor is more efficient in more severe disease is consistent with recent data showing that hepatic PPARα expression is reduced in advanced inflammatory and fibrotic NASH and that resolution of NASH is associated with a recovery of PPARα expression.31 Whatever the explanation for the failure of elafibranor to significantly outperform placebo in patients with mild disease, it is important to note that these patients are usually not considered eligible for pharmacologic therapy, but rather should be managed through dietary and lifestyle changes. The 2 previous large trials in NASH that had NAS reduction as a primary outcome, only included patients with an NAS ≥4,11, 21 and current practice for drug development is to include only patients with moderate or severe disease, defined by a NAS ≥4. Similarly, it has been shown that fibrosis is a strong predictor of liver-related deaths7 and patients with fibrosis are at highest need for pharmacotherapy. In secondary analyses of patients with moderate or severe NASH, 120 mg elafibranor was better than placebo, regardless of the presence or severity of fibrosis. The histologic benefit of the 120-mg dose was mirrored by a significant improvement in liver function tests, in particular alanine aminotransferase, γ-glutamyltransferase, and alkaline phosphatase, and in noninvasive serum panels of steatosis (Steatotest, Fatty Liver Index) and fibrosis (NAFLD Fibrosis score and Fibrotest), which are likely more sensitive and earlier response indicators than histology.
In order to randomize 270 patients, 56 sites were selected, with competitive recruitment and centralized randomization. Due to the unexpectedly high recruitment rates and the staggered design, treatment distribution across the sites was imbalanced. Patient recruitment ranged from 1 to 24 randomized patients per site, and only 15 sites had patients randomized in all 3 treatment arms. In an exploratory, post-hoc analysis designed to control for both center effect and baseline severity, the efficacy of 120 mg elafibranor was explored in the subset of patients with bNAS ≥4 from centers that randomized at least 1 patient per treatment arm. Both NASH resolution and a reduction by ≥2 points in the NAS were achieved more often than placebo. Interestingly, the 21% response rate of the placebo arm for a 2-point NAS reduction is comparable to previous studies,11, 21 thus suggesting that this subgroup of the population is representative of patients included in previous trials.
Because prevention of the occurrence of cirrhosis is the ultimate goal, both from a clinical and a regulatory standpoint,26 drug therapies for NASH should ideally impede fibrogenesis, either directly or indirectly, as a consequence of clearing steatohepatitis. Fibrosis reduction has been an elusive goal so far,11, 32 but recently, a randomized trial of obeticholic acid reported a reduction in fibrosis stage over 18 months of therapy in NASH patients.21 The GOLDEN-505 trial was shorter and not designed for anti-fibrotic end points. It provided the proof-of-principle that resolution of steatohepatitis can result in improvement of fibrosis, an indirect anti-fibrotic effect. Responders for the primary end point, at the 120-mg elafibranor dose, experienced a significant reduction in fibrosis, which was not seen in the overall group of treated patients. Whether a direct anti-fibrotic potency of elafibranor, reported in experimental murine models of fibrosis,16 can be reproduced in humans deserves specific testing in longer trials. Future phase 3 trials will evaluate the effect of elafibranor on the rate of progression to cirrhosis as a result of the resolution of NASH or also through a direct anti-fibrotic effect.
An equally important aspect when treating patients with NASH is the requirement for absence of deterioration (or at best improvement) of the cardiometabolic comorbidities that contribute to overall mortality.25, 26 In addition, insulin resistance, an almost constant feature of NASH, could be causally related to the hepatic buildup of fat, induction of lipotoxic compounds within the liver, and systemic and adipose tissue inflammation. All of these pathways contribute to liver injury and fibrosis and, therefore, improving insulin sensitivity could also have beneficial effects on hepatic damage, as trials of pioglitazone have shown.9, 10, 11 As expected from earlier phase 2 studies,17, 18 including a hyperinsulinemic-euglycemic clamp study in insulin-resistant patients, elafibranor improved markers of insulin resistance, such as the HOMA-IR index, hyperinsulinemia, and free fatty acids, and also significantly reduced HbA1c in diabetics, which reflects improved glycemic control. The pro-atherogenic lipid profile of NASH patients was also improved with significant reductions of total and low-density lipoprotein cholesterol, and increases in HDL cholesterol at both elafibranor doses. Remarkably, the improvements in glycemic and lipid parameters were achieved in patients already treated with conventional glucose and lipid-lowering therapies, which suggests an additional, direct effect of PPARα/δ agonism. It is interesting to note that contrary to placebo-induced resolution of NASH, patients that met the primary end point on elafibranor also exhibited a greater degree of improvement of metabolic and inflammatory parameters than nonresponders. The temporal interaction and dose dependency between the metabolic effects and the histologic response of elafibranor remains to be elucidated in larger trials.
Elafibranor showed a very good tolerability and safety profile throughout the 1-year exposure in this trial. This is of paramount importance, as NASH therapies are expected to be taken on a long-term basis. In addition, these patients often have asymptomatic liver disease and are therefore less willing to tolerate drug-induced side effects in the long term. There was a mild, isolated, and reversible increase in creatinine levels in some patients, and longer post-treatment follow-up is necessary to confirm the reversibility of this biologic effect. PPARα agonists, such as fenofibrate, are known to induce reversible increases in serum creatinine without promoting renal failure, as a result of a pharmacodynamic effect.33, 34, 35 The mechanisms are not entirely known, but might involve increased skeletal muscle production. An improvement in renal function upon fibrate treatment has been reported in a meta-analysis36that described a reduction in albuminuria progression. Here, the increase in creatinine was lower than that observed with fenofibrate (7.1% with 120 mg elafibranor vs 17.2% with fenofibrate; a >20% increase in half of the treated population from the ACCORD [Action to Control Cardiovascular Risk in Diabetes] trial34). However, the absence of an adverse effect of elafibranor on renal function in patients with NASH should be confirmed in larger trials.
This trial has several other strengths and some limitations. The rigorous centralized pathologic reading for both inclusion and end-of-treatment biopsies avoided inclusion of patients without clearly defined NASH,24 and provided uniformity and lack of inter-observer variability for the assessment of histologic end points. The proportion of screen failures for histology was low, thus ensuring that included patients were representative of most real-life NASH patients seen in tertiary centers. Likewise, the low proportion of missing end-of-treatment biopsies minimized potential biases due to patient retention. Another strength is that this was the first large, international, multicenter trial in NASH. However, there were also methodologic limitations. The staggered design of the trial could have resulted in unequal access to the 3 treatment arms, as the randomization sequence was not set upfront for the 3 arms. The competitive recruitment resulted in a variable number of included patients in each center and in an uneven distribution between treatment arms that contributed to a significant center effect. Finally, the inclusion of patients with mild steatohepatitis (bNAS 3) might have blunted the effect of elafibranor in the overall ITT population, as it resulted in a high placebo response rate. Nonetheless, the size of the trial allowed exploratory subgroup analyses that strengthened the demonstration of efficacy. Although secondary analyses are to be considered with caution, this was not a registration, phase 3 trial, but a proof-of-concept, phase 2b exploratory trial designed to inform the design of subsequent, larger, pivotal studies.
The results of this trial compare favorably with results of other investigational agents tested in comparable trials. For instance in the FLINT (Farnesoid X Receptor (FXR) Ligand Obeticholic Acid in NASH Treatment) trial, obeticholic acid induced resolution of NASH in 22% of patients vs 13% in the placebo group. The difference became significant in a post-hoc analysis of the subset of patients with well-defined steatohepatitis at baseline: 19% vs 8%, respectively (P < .05). These rates of response are very close to those obtained in the current trial in the ITT population and in the subset with NAS ≥ 4 (all FLINT participants had an NAS ≥ 4). In post-hoc analyses from the subgroup of patients with well-defined NASH in the PIVENS (Pioglitazone vs Vitamin E vs Placebo for Treatment of Non-Diabetic Patients With Nonalcoholic Steatohepatitis) trial, pioglitazone induced resolution of steatohepatitis in 47% of patients (21% for placebo; P = .001) and vitamin E in 36% (P = .05). - [from Jules: pioglitazone has bad bone affects] - Direct comparisons between molecules are misleading in the absence of head-to-head trials, and because of differences in inclusion criteria and in definitions of histologic response. Importantly, a detailed definition of "resolution of steatohepatitis" is not available from FLINT or PIVENS. In addition, there was no requirement for the absence of "worsening of fibrosis" when defining resolution of NASH as an end point for either PIVENS or FLINT, which further limits comparisons between rates of response with the GOLDEN-505 trial. Only large, phase 3 trials will provide reliable estimates of treatment response for obeticholic acid and elafibranor, but what is clear so far is that a majority of patients are nonresponders and that additional pharmacologic strategies will be necessary to optimize the response rate.
In conclusion, this randomized controlled trial provides evidence that pharmacologic modulation of the PPARα/δ nuclear receptors results in substantial histologic improvement in NASH, including resolution of steatohepatitis and improvement of the cardiometabolic risk profile, with a favorable safety profile. Larger phase 3 trials of elafibranor in the target population of patients with moderate to severe NASH are warranted.
Methods
Study Design
This international, multicenter, randomized placebo-controlled study tested elafibranor at the dose of 80 mg and 120 mg once a day vs placebo over 52 weeks and was conducted at 56 sites, 19 in the United States and 37 in 8 European countries. The study had a staggered design as requested by the regulatory agencies to test the safety of elafibranor during a 6-month period at the lower dose before exposing patients for 1 year at the highest dose. During the first recruitment phase, 172 patients were screened between September 2012 and June 2013 for treatment with 80 mg/d of elafibranor or placebo (allocation 2:1). The second recruitment period at the dose of 120 mg/d started in July 2013, when 179 patients were screened in 1 week. The randomization of this second cohort started in October 2013 (allocation of elafibranor 120 mg or placebo in a 2:1 ratio), after unrestricted approval from the Independent Data and Safety Monitoring Board. The clinical study protocol was approved in all countries by National Authorities and Ethics Committees. All patients gave written informed consent. All authors had access to the study data and have approved and reviewed the final manuscript.
Patients
The inclusion criteria included age 18 to 75 years and a histologic diagnosis of noncirrhotic NASH confirmed by a central pathologist. Patients were excluded if daily alcohol consumption was more than 2 drink units/d (equivalent to 20 g) in women and 3 drink units/d (30 g) in men, if steatohepatitis was due to secondary causes, or if any other chronic liver disease was identified.
Randomization and Masking
Randomization was obtained through a computer-generated coding list, and treatment allocation was performed centrally for all sites through a web system, based on date of randomization, and stratified for diabetes. No stratification was made on investigation sites. Elafibranor and placebo were provided as identical capsules in wallets labeled with code numbers. Patients, investigators, clinical site staff, and the pathologist were masked to treatment assignment. The allocation of treatment was done in a 1:1:1 ratio for the 3 treatment arms-placebo, elafibranor 80 mg, and elafibranor 120 mg.
Procedures
Patients were followed every 2 months with clinical and laboratory evaluations throughout the 1-year treatment period. An end-of-treatment biopsy and a 3-month post-treatment follow-up visit were performed. Screening and end-of-treatment biopsies were all read centrally by a single pathologist in a blinded manner (PB). At end of study, all slides (baseline and end of study) were read in scrambled order. For inclusion, the liver biopsy needed to be collected within the past 9 months. Steatohepatitis was diagnosed based on the presence of steatosis (>5% of hepatocytes), hepatocyte ballooning, and lobular inflammation. Fibrosis was evaluated using the NASH CRN fibrosis staging system. Included patients had an NAFLD activity score (NAS) ranging from 3 to 8, with at least 1 for steatosis, ballooning, and inflammation. All stages of fibrosis (0-3) were accepted, except for cirrhosis. Noninvasive panels for steatosis or fibrosis (Fatty Liver Index, SteatoTest, Fibrotest, and the NAFLD Fibrosis score) were measured at baseline, 6 months, and 12 months (end of treatment). Biologic assessments were all centralized and performed at each visit for efficacy and safety purposes.
Outcomes
The primary outcome was reversal of NASH without worsening of fibrosis. This was defined as per protocol, before study start, as the absence (score of 0) of at least 1 of the 3 components of NASH, that is, steatosis, ballooning, and inflammation; worsening of fibrosis was defined as the progression to bridging fibrosis (ie, stage 3) or cirrhosis in patients without bridging fibrosis at baseline or to cirrhosis in patients with bridging fibrosis at baseline.
After the study was completed, a modified and more stringent definition was proposed by academic and regulatory experts and recommended by regulatory agencies for ongoing trials.19, 20 It defines resolution of NASH as disappearance of ballooning (score = 0), together with either disappearance of lobular inflammation or the persistence of mild lobular inflammation only (score = 0 or 1), and resulting in an overall pathologic diagnosis of either steatosis alone or steatosis with mild inflammation; any stage increase in fibrosis is considered fibrosis progression. Because this more stringent definition is now used for current and future trials, we will here report on both the protocol-defined and the post-hoc analysis of the modified definition.
Secondary outcomes included changes in NAS between end of treatment and baseline biopsy (including the proportion of patients with a 2-point decrease); changes and improvements in individual histologic scores of steatosis, ballooning, inflammation, and fibrosis; changes in liver enzymes, in noninvasive markers of steatosis and fibrosis, in lipid and glycemic parameters, in surrogate markers of insulin resistance (fasting insulin and Homeostasis Model Assessment Index scores); changes in systemic inflammatory markers; and safety and tolerability of elafibranor at both doses.
Statistical Methods
The main selection was the population of all randomized patients that received at least one dose of study drug (intention-to-treat [ITT] sample). To assess the robustness of findings, sensitivity analyses were performed using the per-protocol population defined as the ITT population with available liver biopsy at the end of the study. For sensitivity purposes, the following post-hoc selections were considered: patients with baseline NAS (bNAS) ≥4 (moderate or high disease activity), who are similar to those included in previous NASH trials11, 21; patients with bNAS ≥4 and fibrosis of any stage at baseline; patients with bNAS ≥4 and fibrosis stage ≥2 at baseline (target patient population for current phase 3 trials), and patients with bNAS ≥4 recruited in centers that randomized at least 1 patient in each treatment arm (justified by the strong treatment-center imbalance). The ITT population was the main selection and a significant effect observed in the ITT population was conditional to test the significance in the other subpopulations.
The main analysis was a mixed model featuring logistic regression on therapy response with treatment as fixed factor (placebo, 80 mg, and 120 mg), adjusted for bNAS. The multicenter context was accounted for by random factor. Based on the assumption of superiority of the 120-mg dose, testing the 80-mg dose was conditional to the significance of the effect of 120 mg (step-down testing22). No multiplicity correction was needed due to step-down strategy.22 For patients with liver biopsy unavailable at the end of treatment, a worst-case imputation in assimilating missing value to therapy failure was considered.
Post-hoc analyses tested the main treatment effect and its interaction with baseline severity (bNAS).
For easier clinical discussion, risk ratio was reported with odds ratio (OR) derived from logistic regression. Geometric mean change over baseline and related t tests were used to compare the treatment subgroups on biologic parameters, composite biomarker scores for NAFLD and fibrosis.
For sample size calculations, we assumed a 20% and 45% responder rate in the placebo and 120-mg dose groups, respectively, and a dropout rate of 25%. Ninety patients per group were required to reach this difference, with a power of 80% at a 2-sided .05 significance test level. The analyses were conducted with the Statistical Package R (release 3.1.1), all tests were conducted at .05 two-sided level.
Role of the Funding Source
The GOLDEN-505 study was sponsored by Genfit SA. The protocol was written by a panel of academic experts and sponsor representatives and amended in accordance to input from regulatory bodies. The corresponding author had full access to all the data in the study and had final responsibility for manuscript submission.
Results
A total of 276 patients were randomized, 92 in the placebo group, 93 in the elafibranor 80 mg group, and 91 in the elafibranor 120-mg group (Figure 1). Two patients did not receive the study medication and the remaining 274 patients constitute the ITT population. Thirty-three patients (12%) dropped out during the study (Supplementary Table 1). Final liver biopsies were available in 237 patients (77, 82, and 78 patients in the placebo, elafibranor 80 mg, and elafibranor 120 mg groups, respectively). Of these, only 5 patients were no longer diagnosed as having NASH on the baseline biopsy upon scrambled re-reading at end of study. This did not modify the overall results.
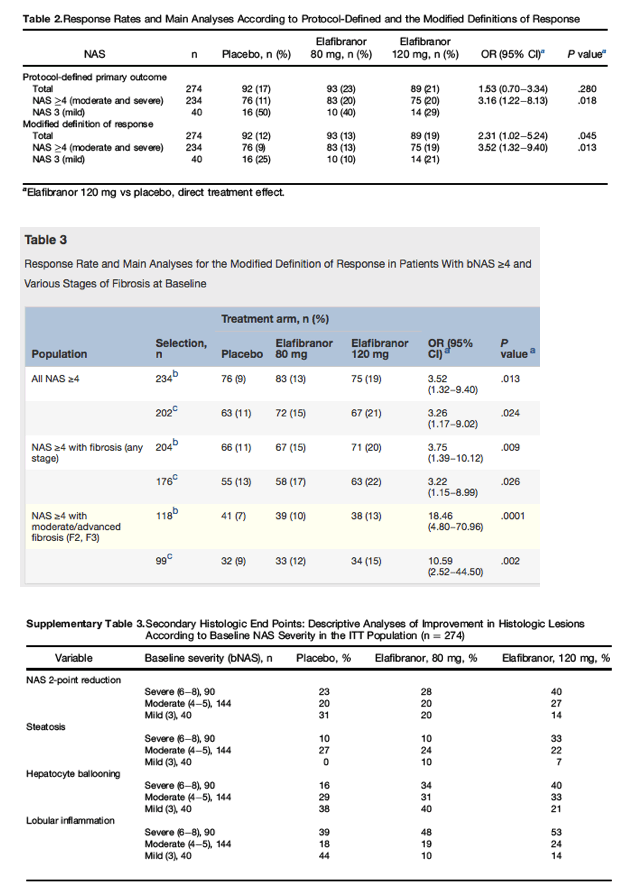
Table 2 shows the response rates and corresponding risk ratios in the ITT population for the primary outcome. There was no difference between the elafibranor arms and placebo according to the protocol-defined definition. A post-hoc analysis using the modified definition of response shows that the response rate was significantly higher for the 120-mg arm than for placebo (19% vs 12%; OR = 2.31; 95% confidence interval [CI]: 1.02-5.24; P = .045). The 80-mg arm did not perform better than placebo for both definitions of response, the protocol-based and the modified definition (OR = 1.48; 95% CI: 0.7-3.14; P = .30 and 1.11, 95% CI: 0.48-2.57; P = .80, respectively), or for any other histologic analysis.
Results of the secondary histologic outcomes (Supplementary Table 3) show no significant difference between elafibranor and placebo. Nonetheless, the efficacy of the 120-mg dose to reduce the NAS by 2 points and to improve steatosis, ballooning, and lobular inflammation was more pronounced with increasing baseline severity, in contrast to the absence of a clear pattern in the placebo or 80-mg groups.
A number of post-hoc, secondary analyses were performed. Importantly, there was a strong interaction effect between baseline severity and elafibranor dose, which was significant for 120 mg for both the protocol-defined (OR = 2.63; 95% CI: 1.25-5.52; P = .012) and modified definition (OR = 2.76; 95% CI: 1.33-5.76; P = .007) (Supplementary Table 2). The significant interaction effect with baseline severity indicated that the efficacy of elafibranor 120 mg vs placebo increased with baseline severity. Hence, the exclusion of patients with mild disease activity (bNAS = 3, n = 40) revealed a significant direct effect of elafibranor 120 mg vs placebo (OR = 3.16; 95% CI: 1.22-8.13 and OR = 3.52; 95% CI: 1.32-9.40, for the protocol-defined and modified definitions, respectively) in the remaining population of 234 patients with bNAS ≥4 (85% of the ITT population); there was no significant difference for the 80-mg arm. Overall, the 120-mg elafibranor dose doubled the proportion of responders vs placebo in patients with bNAS ≥4.
As patient recruitment was based on a wide spectrum of baseline severity (NAS 3-8 and fibrosis stage 0-3), we performed post-hoc analyses in NAS ≥4 populations with increasing fibrosis stages (Table 3). The response rates of 120 mg elafibranor for the protocol-defined definition were significantly higher than that of placebo, while there was no significant difference for the 80-mg arm. The 120-mg dose was also more effective in the subpopulation of patients with any fibrosis (F1-F3), as well as in those with moderate or advanced fibrosis (F2-F3) (Table 3). The results were qualitatively similar when using the modified definition (data not shown).
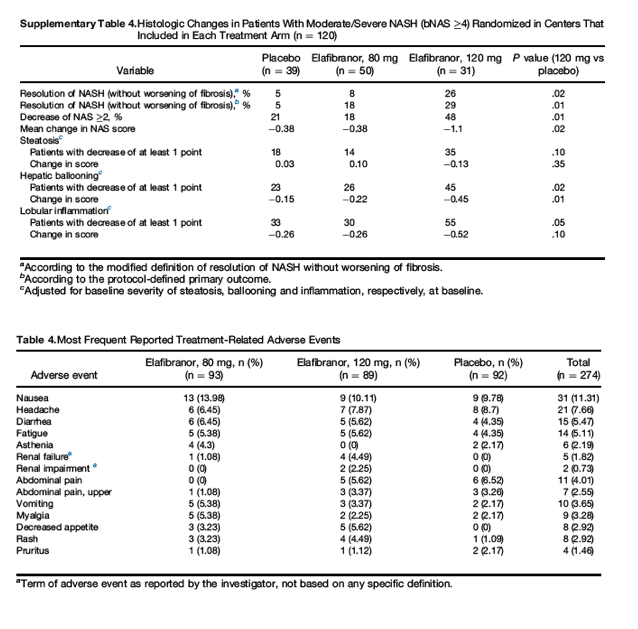
Because of a heterogeneous center effect and the unbalanced treatment-center distribution (due to the staggered design and an unexpected high rate of recruitment), we performed an analysis in the subset of bNAS ≥4 patients recruited in centers that randomized at least 1 patient in each treatment arm (n = 120, Supplementary Table 4). The response rates were 29% and 26% (protocol-defined and modified definitions) vs 5% placebo (P = .01 and.02, respectively). Forty-eight percent of patients improved the NAS by ≥2 points (vs 21% in the placebo arm; P = .013). Hepatocyte ballooning and lobular inflammation were also significantly improved, with a trend toward improvement in steatosis but not fibrosis.
Finally, we tested whether patients that achieved resolution of NASH without worsening of fibrosis in the 120-mg elafibranor arm also experienced improvement in fibrosis.
Supplementary Figure 1 shows strong reductions in fibrosis, hepatocyte ballooning, and the NAS (all P < .001), as well as in lobular inflammation and steatosis (both P < .05), when compared with nonresponders to the same regimen. These findings were similar with both definitions of response.
Patients treated with both elafibranor doses (80 mg and 120 mg) improved liver function tests (alanine aminotransferase, γ-glutamyltransferase, and alkaline phosphatase; Figure 2A-C) and lipid parameters (triglycerides, low-density lipoprotein cholesterol, HDL cholesterol, Figure 2D-F). In diabetic patients (40% of the Intention to Treat population), elafibranor improved fasting serum glucose (-0.98 ± 0.56 mmol/L for 120 mg vs placebo; P = .08) and HbA1c (-0.46% for 120 mg vs placebo; P = .038), as well as markers of insulin resistance (fasting insulin, HOMA-IR, and circulating free fatty acids, Figure 3). There was a clear reduction in systemic inflammatory markers, such as high-sensitivity C-reactive protein (-42% for 120 mg vs placebo; P = .161), fibrinogen, and haptoglobin at both doses (Supplementary Figure 2A). In line with the histologic changes, serum panel biomarkers of steatosis and fibrosis, such as SteatoTest, Fatty Liver Index, Fibrotest/FibroSure, and the NAFLD Fibrosis score, showed significant reductions in patients treated with elafibranor 120 mg compared with placebo (Supplementary Figure 2B).
Elafibranor was safe and well tolerated. Clinical adverse events were mostly mild and similar in the placebo and elafibranor arms (Table 4). There were no cardiovascular events or deaths in the elafibranor arms. Six patients (6.5%) were discontinued for adverse events in the placebo group, 7 (7.9%) in the 80-mg group, and 5 (5.4%) in the 120-mg groups. There was a mild, reversible but statistically significant increase in serum creatinine (effect size vs placebo: 4.31 ± 1.19 μmol/L; P < .001). Other renal markers, such as cystatin C and microalbuminuria, remained normal. The increase in creatinine led to a reported renal impairment/failure in 7 patients treated with elafibranor (Supplementary Table 5). All of them had increased creatinine at baseline; 1 of them had significant pre-existing increases in creatinine, cystatin C, urinary neutrophil gelatinase-associated lipocalin, urinary creatinine, serum albumin, and urinary albumin; and decreased creatinine clearance, and was therefore discontinued. Weight did not change and there was no significant reduction in hematocrit or hemoglobin vs placebo. Serious adverse events occurred in 11 patients in the placebo (12%), 15 patients in the 80-mg (16.1%), and 14 patients in the 120-mg (15.8%) arms. Treatment-related serious adverse events occurred in 2 patients in the 80-mg elafibranor arm (spontaneous abortion, ataxia, fasciculation, and tremor), in 2 patients in the elafibranor 120 mg arm (acute pancreatitis, Parkinson disease), and in 4 patients from the placebo arm (renal cancer, breast cancer, bladder cancer, and pancreatic cancer).
Neoplastic serious adverse events were reported in 6 patients during the study and the 3-month follow-up periods: one bladder cancer in the elafibranor 80-mg arm (unlikely related to study drug) in a patient with previous doubtful cytologic lesions, and 5 cancers in the placebo arm (the 4 described above and 1 esophageal cancer considered unlikely related to study drug).
|
| |
|
|
|
|
|