| |
Efficacy of 8 Weeks of Sofosbuvir, Velpatasvir, and Voxilaprevir in Patients With Chronic HCV Infection: 2 Phase 3 Randomized Trials
|
| |
| |
Download the PDF here
Download the PDF here
Download the PDF here
Gastroenterology April 5 2017 - Ira M. Jacobson,1, Eric Lawitz,2 Edward J. Gane,3 Bernard E. Willems,4 Peter J. Ruane,5 Ronald G. Nahass,6 Sergio M. Borgia,7 Stephen D. Shafran,8 Kimberly A. Workowski,9 Brian Pearlman,10 Robert H. Hyland,11 Luisa M. Stamm,11 Evguenia Svarovskaia,11 Hadas Dvory-Sobol,11 Yanni Zhu,11 G. Mani Subramanian,11 Diana M. Brainard,11 John G. McHutchison,11 Norbert Bräu,12,13 Thomas Berg,14 Kosh Agarwal,15 Bal Raj Bhandari,16 Mitchell Davis,17 Jordan J. Feld,18 Gregory J. Dore,19 Catherine A. M. Stedman,20 Alexander J. Thompson,21 Tarik Asselah,22 Stuart K. Roberts,23 and Graham R. Foster24,
Background & Aims
Patients with chronic hepatitis C virus (HCV) infection have high rates of sustained virologic response (SVR) after 12 weeks of treatment with the nucleotide polymerase inhibitor sofosbuvir combined with the NS5A inhibitor velpatasvir. We assessed the efficacy of 8 weeks of treatment with sofosbuvir and velpatasvir plus the pangenotypic NS3/4A protease inhibitor voxilaprevir (sofosbuvir-velpatasvir-voxilaprevir).
Methods
In 2 phase 3, open-label trials, patients with HCV infection who had not been treated previously with a direct-acting antiviral agent were assigned randomly to groups given sofosbuvir-velpatasvir-voxilaprevir for 8 weeks or sofosbuvir-velpatasvir for 12 weeks. POLARIS-2, which enrolled patients infected with all HCV genotypes with or without cirrhosis, except patients with genotype 3 and cirrhosis, was designed to test the noninferiority of 8 weeks of sofosbuvir-velpatasvir-voxilaprevir to 12 weeks of sofosbuvir-velpatasvir using a noninferiority margin of 5%. POLARIS-3, which enrolled patients infected with HCV genotype 3 who had cirrhosis, compared rates of SVR in both groups with a performance goal of 83%.
Results
In POLARIS-2, 95% (95% confidence interval [CI], 93%-97%) of patients had an SVR to 8 weeks of sofosbuvir-velpatasvir-voxilaprevir; this did not meet the criterion to establish noninferiority to 12 weeks of sofosbuvir-velpatasvir, which produced an SVR in 98% of patients (95% CI, 96%-99%; difference in the stratum-adjusted Mantel-Haenszel proportions of 3.4%; 95% CI, -6.2% to -0.6%). The difference in the efficacy was owing primarily to a lower rate of SVR (92%) among patients with HCV genotype 1a infection receiving 8 weeks of sofosbuvir-velpatasvir-voxilaprevir. In POLARIS-3, 96% of patients (95% CI, 91%-99%) achieved an SVR in both treatment groups, which was significantly superior to the performance goal. Overall, the most common adverse events were headache, fatigue, diarrhea, and nausea; diarrhea and nausea were reported more frequently by patients receiving voxilaprevir. In both trials, the proportion of patients who discontinued treatment because of adverse events was low (range, 0%-1%).
Conclusions
In phase 3 trials of patients with HCV infection, we did not establish that sofosbuvir-velpatasvir-voxilaprevir for 8 weeks was noninferior to sofosbuvir-velpatasvir for 12 weeks, but the 2 regimens had similar rates of SVR in patients with HCV genotype 3 and cirrhosis. Mild gastrointestinal adverse events were associated with treatment regimens that included voxilaprevir.
Since the introduction of drugs that directly interfere with the replication of the hepatitis C virus (HCV), the treatment of patients with chronic HCV infection has improved steadily.1, 2, 3 Combinations of direct-acting antiviral agents (DAAs) with complementary mechanisms of action have brought rates of virologic failure down to 5% or less in most patient populations. Pangenotypic, single-tablet, all-oral regimens have improved the simplicity and convenience of dosing.4 Although the duration of standard treatment has been reduced from 24 to 48 weeks with interferon-containing regimens to 12 weeks with DAA regimens, the feasibility of further shortening the duration of treatment without loss of efficacy is being explored actively in all patient populations, including those who have been considered difficult to treat, such as patients with genotype 3 infection and cirrhosis.5, 6, 7 Potential approaches to shortening treatment duration include developing novel double and triple DAA combination therapies. A possible benefit of shortened treatment duration is increased patient adherence, which may in turn improve efficacy.6, 8
Sofosbuvir is a nucleotide analog HCV NS5B polymerase inhibitor that, in combination with other agents, has been approved for the treatment of HCV.9, 10, 11 Velpatasvir is an HCV-NS5A inhibitor with pangenotypic potency.12 Twelve weeks of the fixed-dose combination of sofosbuvir and velpatasvir, which has been approved for the treatment of patients with HCV infection of all genotypes, provided high levels of sustained virologic response (SVR) in phase 3 clinical trials in both treatment-naive and treatment-experienced patients.13, 14 Voxilaprevir (formerly GS-9857; Gilead Sciences, Foster City, CA) is a macrocyclic, pangenotypic inhibitor of the NS3/4A protease with an improved resistance profile in comparison with earlier protease inhibitors.15, 16, 17 In phase 2 trials, 8 weeks of treatment with the combination of sofosbuvir, velpatasvir, and voxilaprevir provided high levels of SVR in patients chronically infected with HCV of all genotypes who had not received treatment with direct-acting antivirals previously.18, 19, 20, 21
We conducted 2 phase 3 trials designed to assess the efficacy and safety of 8 weeks of treatment with the fixed-dose combination of sofosbuvir, velpatasvir, and voxilaprevir in patients chronically infected with HCV of any genotype, including patients with compensated cirrhosis, who had not received treatment previously with direct-acting antivirals.
Discussion
In these international phase 3 trials, treatment with sofosbuvir-velpatasvir-voxilaprevir for 8 weeks resulted in high rates of SVR in HCV-infected patients with and without compensated cirrhosis who had not received treatment previously with DAAs. However, in the POLARIS-2 trial, the rate of SVR of 95% in patients receiving sofosbuvir-velpatasvir-voxilaprevir for 8 weeks was not shown to be noninferior to that of 98% in patients receiving sofosbuvir-velpatasvir for 12 weeks. The difference in the efficacy primarily was owing to the higher rate of relapse among patients with genotype 1a HCV infection receiving 8 weeks of sofosbuvir-velpatasvir-voxilaprevir. Among patients receiving sofosbuvir-velpatasvir-voxilaprevir for 8 weeks with HCV genotypes other than genotype 1a, the rate of SVR was 97%. In the POLARIS-3 trial, which enrolled only the most challenging patient population—patients with genotype 3 and cirrhosis—very high and identical rates of SVR (96%), and low rates of virologic failure (2%) were observed in both the 8- and 12-week treatment groups.
The lower rate of SVR among patients with HCV genotype 1a as compared with patients with other genotypes receiving 8 weeks of sofosbuvir-velpatasvir-voxilaprevir was an unexpected finding. This difference was not observed in the phase 2 clinical trials, which enrolled a similar but smaller patient population, underscoring the importance of conducting large, randomized, controlled clinical trials before investigational drug approval.19, 22 Among patients with HCV genotype 1a, the rate of SVR was 89% in those who had NS3 or NS5A resistance-associated substitutions at baseline and 95% among those who did not, with most of the substitutions being at position 80 in the NS3 protein. Although the Q80K polymorphism, which occurs commonly in patients with genotype 1a, indeed was associated with lower rates of SVR in POLARIS-2, the causal significance of this finding is unclear; in contrast to its effect on susceptibility to simeprevir, another HCV protease inhibitor, this substitution has not been found to alter in vitro susceptibility to voxilaprevir.22, 23, 24 Furthermore, Q80K was not observed to emerge at the time of failure among the HCV genotype 1a patients with relapse and was no longer detectable in 1 patient at relapse despite its presence before treatment. The emergence of resistance-associated substitutions was uncommon overall among patients who did not achieve SVR after 8 weeks of sofosbuvir-velpatasvir-voxilaprevir, consistent with the high barrier to resistance for this regimen and suggesting that virologic failure was driven predominantly by insufficient treatment duration to eliminate the virus.25, 26
In the POLARIS-2 and POLARIS-3 trials, treatment for 8 weeks with sofosbuvir-velpatasvir-voxilaprevir led to high rates of SVR in patients with genotype 3 infection. These high rates of cure are comparable with those observed with 12 weeks of sofosbuvir-velpatasvir in the current studies, as well as those seen in a prior phase 3 trial,14 and support ribavirin-free regimens, even for those with compensated cirrhosis, who are considered more difficult to cure.
The rates of adverse events in the patients receiving sofosbuvir-velpatasvir-voxilaprevir generally were similar to the rates among patients receiving sofosbuvir-velpatasvir, except that more patients receiving sofosbuvir-velpatasvir-voxilaprevir had mild nausea and diarrhea, which has been observed with prior NS3/4A protease inhibitors.27 No patient interrupted treatment or discontinued early as a result of these events. There was no evidence of voxilaprevir-related hepatotoxicity.
In the era of highly effective and well-tolerated DAA-based regimens, nonadherence has become the most important risk factor for treatment failure.
Sofosbuvir-velpatasvir-voxilaprevir provides a highly efficacious and well-tolerated short-duration regimen for the treatment of HCV in patients for whom adherence to a longer-duration regimen may be challenging. As the population of patients being treated for HCV expands from those engaged in health care systems to marginalized populations of homeless, incarcerated, or those with addictions, the availability of a highly effective 8-week treatment regimen may enable more HCV-infected patients to be treated successfully with less burden on the resources of patients and providers. This combination also may provide important benefits in regions (eg, Europe) or communities (eg, injection drug users) where the prevalence of HCV genotype 3 is comparatively high.
Materials and Methods
Patients
Both trials enrolled patients who had not received treatment previously for HCV infection with regimens containing DAAs. The POLARIS-2 trial enrolled patients of all HCV genotypes with and without compensated cirrhosis, except patients with HCV genotype 3 who were enrolled only if they did not have cirrhosis. The POLARIS-3 trial enrolled patients with HCV genotype 3 and cirrhosis exclusively. Eligibility criteria otherwise were identical for the 2 trials. Full eligibility criteria for both trials are provided in the Supplement.
Study Designs
In both of these multicenter, randomized, open-label trials, patients received either a fixed-dose combination tablet containing 400 mg of sofosbuvir, 100 mg of velpatasvir, and 100 mg of voxilaprevir once daily for 8 weeks, or a fixed-dose combination tablet containing 400 mg of sofosbuvir and 100 mg of velpatasvir once daily for 12 weeks.
In POLARIS-2, patients were enrolled at 117 sites in the United States, Canada, New Zealand, Australia, France, Germany, and the United Kingdom from November 2015 through April 2016. For HCV genotypes 1, 2, and 4, enrollment of at least 30% of patients with compensated cirrhosis was targeted per genotype. Patient randomization was managed by using an interactive web response system (Bracket). A statistician employed by the sponsor (L.H.) generated the randomization code using the SAS program, which was validated by another statistician employed by the sponsor. Patients with genotypes 1, 2, 3, and 4 were randomized at a 1:1 ratio stratified by 3 factors (genotype, cirrhosis status, and treatment history), and a block size of 4 to receive sofosbuvir-velpatasvir-voxilaprevir for 8 weeks or sofosbuvir-velpatasvir for 12 weeks. All patients with other HCV genotypes, with or without cirrhosis, were enrolled in the sofosbuvir-velpatasvir-voxilaprevir group.
POLARIS-3 was a separate dedicated study for patients with genotype 3 and compensated cirrhosis who were excluded from POLARIS-2. Patients were enrolled at 84 sites in the United States, Canada, New Zealand, Australia, France, Germany, and the United Kingdom from January 2016 through April 2016. As with POLARIS-2, patient randomization was managed by using an interactive web response system (Bracket). Patients were randomized in a 1:1 ratio stratified by history of prior treatment with interferon with a block size of 4 to receive either sofosbuvir-velpatasvir-voxilaprevir for 8 weeks or sofosbuvir-velpatasvir for 12 weeks.
Assessments
For both trials, screening assessments included measurement of the serum HCV-RNA level, interleukin 28B genotyping, and standard laboratory and clinical tests. HCV-RNA levels were measured using the COBAS AmpliPrep/COBAS TaqMan HCV Quantitative Test, version 2.0, with a lower limit of quantification of 15 IU/mL. The presence of cirrhosis was determined on liver biopsy showing cirrhosis (Metavir stage 4, or Ishak score of 5 or 6), a FibroTest score of more than 0.75 and a ratio of aspartate aminotransferase to platelets of more than 2, or a FibroScan result of more than 12.5 kPa.
The Abbott RealTime HCV genotype II assay was used to determine HCV genotype at screening. HCV genotype and subtype subsequently were determined by analysis of NS3, NS5A, and NS5B sequences from deep sequencing using the basic local alignment search tool (BLAST). All analyses were conducted using the BLAST genotyping.
Deep sequencing of the HCV NS3, NS5A, and NS5B coding regions was performed on samples obtained from all patients at baseline and again for all patients with virologic failure. We report substitutions associated with resistance to drugs within the classes that were present in more than 15% of the sequence reads.
End Points
For both studies, the primary efficacy end point was SVR (serum HCV-RNA level <15 IU/mL) 12 weeks after the end of treatment in all patients who were enrolled and received at least 1 dose of study drug. Secondary efficacy end points included the kinetics of circulating HCV RNA during and after treatment, and the emergence of viral resistance during and after treatment. The primary safety end point was the proportion of patients who discontinued treatment prematurely owing to adverse events.
Statistical Analysis
In POLARIS-2, the primary efficacy analysis assessed the noninferiority of the rate of SVR among patients receiving sofosbuvir-velpatasvir-voxilaprevir to the rate among patients receiving sofosbuvir-velpatasvir using a noninferiority margin of 5%. A 2-sided 95% confidence interval (CI) was constructed for the difference in the rates of SVR between the 2 treatment groups using stratum-adjusted Mantel-Haenszel proportions. Noninferiority was established if the lower bound was greater than -5%. The planned sample size of 780 patients was calculated to be able to provide more than 95% power to establish noninferiority.
In POLARIS-3, the primary efficacy analysis assessed first the rate of SVR among patients in the sofosbuvir-velpatasvir-voxilaprevir group against a performance goal of 83% using a 2-sided exact 1-sample binomial test at the 0.05 significance level. If this group met this criterion, the rate of SVR in the sofosbuvir-velpatasvir group also would be assessed against the performance goal of 83% at the 0.05 significance level. A sample size of 100 patients was calculated to be able to provide more than 80% power to detect an improvement of 10 percentage points in the rate of SVR. The performance goal of 83% was based on the prior results of sofosbuvir-velpatasvir in this patient population in the ASTRAL-3 trial (SVR, 91%; 95% CI, 83-96).11 All authors had access to the study data and reviewed and approved the final manuscript.
Study Oversight
The 2 studies were approved by the institutional review board or independent ethics committee at each participating site and were conducted in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The studies were designed and conducted by the sponsor (Gilead Sciences) in collaboration with the principal investigators, according to the protocol. The sponsor collected the data, monitored study conduct, and performed the statistical analyses. Independent safety monitoring committees reviewed the progress of the studies. All of the authors had access to the data and assumed responsibility for the integrity and completeness of the reported data. The initial draft of the manuscript was prepared by a writer employed by Gilead Sciences and the primary investigators with input from all of the authors.
Results
Baseline Characteristics
Of the 1116 patients screened for the POLARIS-2 trial, 943 were enrolled and 941 began treatment (Supplementary Table 1). In total, 882 patients with HCV genotypes 1, 2, 3, and 4 were randomized to receive either sofosbuvir-velpatasvir-voxilaprevir for 8 weeks (n = 451) or sofosbuvir-velpatasvir for 12 weeks (n = 431). Forty-eight patients with genotypes 5 and 6, and 2 patients with an unknown genotype, were enrolled to receive sofosbuvir-velpatasvir-voxilaprevir for 8 weeks. Nine patients with HCV genotype 6 by BLAST analysis were identified at screening as having HCV genotype 1 and were assigned to the sofosbuvir-velpatasvir group. Of the 315 patients who were screened for POLARIS-3, 220 were enrolled and 219 began treatment (Supplementary Table 2). Of these, 110 were randomized to the sofosbuvir-velpatasvir-voxilaprevir group, and 109 were randomized to the sofosbuvir-velpatasvir group.
Efficacy
POLARIS-2 trial
In the POLARIS-2 trial, the rate of SVR was 95% (95% CI, 93-97) among patients receiving 8 weeks of sofosbuvir-velpatasvir-voxilaprevir, and 98% (95% CI, 96-99) among those receiving 12 weeks of sofosbuvir-velpatasvir (Table 2), with a difference in the stratum-adjusted Mantel-Haenszel proportions between the 2 groups of -3.4 percentage points (95% CI, -6.2 to -0.6). Given that the lower bound of the 2-sided 95% CI for the difference between the rates of SVR of -6.2% was below the pre-established limit of -5%, 8 weeks of sofosbuvir-velpatasvir-voxilaprevir did not meet the prespecified criteria for noninferiority to 12 weeks of sofosbuvir-velpatasvir (Figure 1).
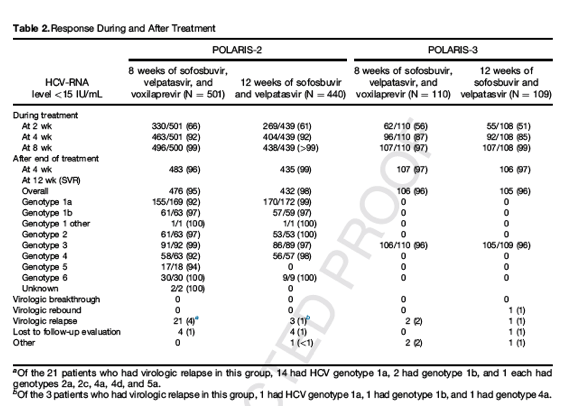
Among patients without cirrhosis, rates of SVR were 96% (394 of 411) for patients receiving sofosbuvir-velpatasvir-voxilaprevir and 98% (349 of 356) for patients receiving sofosbuvir-velpatasvir. Among patients with cirrhosis, 91% (82 of 90) of patients receiving sofosbuvir-velpatasvir-voxilaprevir for 8 weeks had SVR, as compared with 99% (83 of 84) of patients receiving sofosbuvir-velpatasvir for 12 weeks. Among patients with HCV genotype 1a infection, 92% (155 of 169) of patients receiving sofosbuvir-velpatasvir-voxilaprevir for 8 weeks had SVR, as compared with 99% (170 of 172) of patients receiving sofosbuvir-velpatasvir for 12 weeks.
Overall, 21 patients (4%) in the group receiving sofosbuvir-velpatasvir-voxilaprevir had an observed virologic relapse, compared with 3 of 440 (1%) in the group receiving sofosbuvir-velpatasvir. Fourteen of the patients with virologic relapse in the sofosbuvir-velpatasvir-voxilaprevir group were infected with HCV genotype 1a, compared with 1 patient in the sofosbuvir-velpatasvir group. Among patients with HCV genotype 1a without cirrhosis, relapse occurred in 8% and 0% of patients receiving sofosbuvir-velpatasvir-voxilaprevir and sofosbuvir-velpatasvir, respectively, and in 10% and 2% of patients with cirrhosis. No virologic failures occurred in genotype 3 patients. No patients in either treatment group had a virologic breakthrough during treatment. Four patients in each group were lost to follow-up evaluation, including 3 patients with HCV genotype 4 receiving sofosbuvir-velpatasvir-voxilaprevir. These patients with nonvirologic failure in the 8-week group primarily were responsible for the difference in SVR among patients with HCV genotype 4 infection between the 2 groups (92% compared with 98%); of the 120 patients with genotype 4, there were 2 relapses in the sofosbuvir-velpatasvir-voxilaprevir 8-week group and 1 in the sofosbuvir-velpatasvir 12-week group.
POLARIS-3 trial
Among patients with HCV genotype 3 and cirrhosis in the POLARIS-3 trial, the rate of SVR was 96% in both treatment groups (95% CI, 91-99), which was significantly superior to the performance goal of 83% (P < .001 for both groups) (Figure 1). Two patients (2%) in the sofosbuvir-velpatasvir-voxilaprevir group had a virologic relapse by post-treatment week 4. Two patients (2%) in the sofosbuvir-velpatasvir group also had virologic failure: 1 patient who did not achieve on-treatment HCV-RNA suppression had a virologic rebound by week 8 of treatment, and 1 patient had virologic relapse at post-treatment week 4. In both patients, concentrations of GS-331007—the predominant metabolite of sofosbuvir—and velpatasvir were low for at least 1 study visit, suggesting that the patient was not fully adherent to study dosing.
Viral Resistance Testing
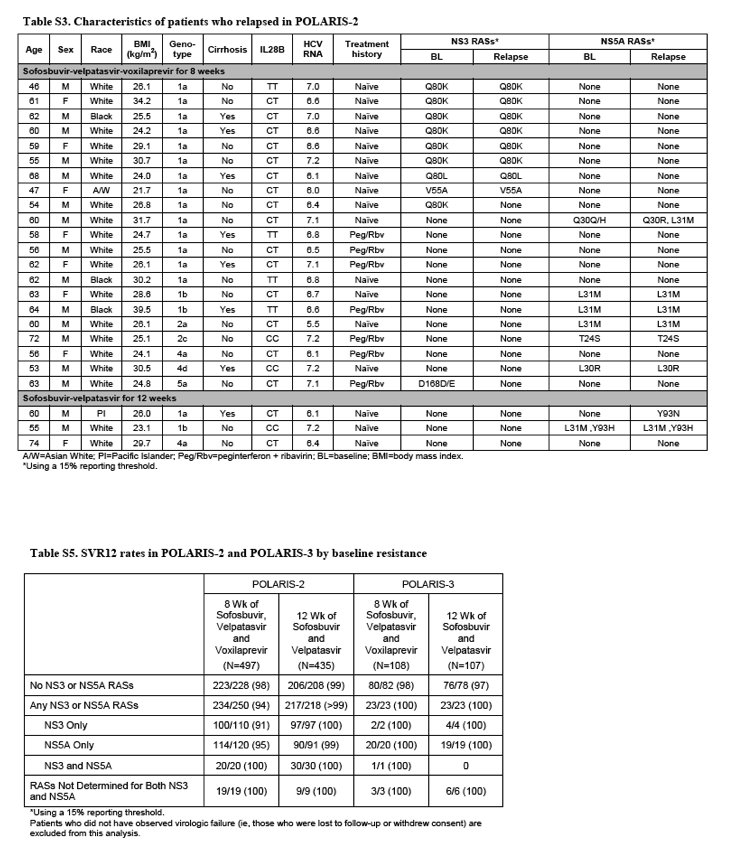
Among the 497 patients receiving sofosbuvir-velpatasvir-voxilaprevir in the POLARIS-2 trial, 250 had viral variants associated with resistance to NS3 and/or NS5A inhibitors at baseline (Supplementary Table 5). Of these, 94% had SVR, as compared with 98% for patients without resistance-associated substitutions at baseline. For patients with HCV genotype 1a, the rates of SVR in patients with and without baseline resistance-associated substitutions were 89% and 95%, respectively. The Q80K resistance-associated substitution was the most commonly observed NS3 variant, although it does not confer a change to voxilaprevir susceptibility in vitro. The Q80K resistance-associated substitution was the most commonly observed NS3 variant; although it confers no change to voxilaprevir susceptibility in vitro,15 baseline Q80K nevertheless was associated with a reduction in SVR rate for genotype 1a patients receiving the sofosbuvir-velpatasvir-voxilaprevir regimen for 8 weeks, 88% with Q80K compared with 94% without. Of the 21 patients who relapsed in the sofosbuvir-velpatasvir-voxilaprevir group, 1 had treatment-emergent NS5A resistance-associated substitutions Q30R and L31M. Among patients receiving sofosbuvir-velpatasvir in POLARIS-2, 1 of the 3 patients who relapsed had the Y93N variant, which is associated with resistance to NS5A inhibitors, at relapse.
In the POLARIS-3 trial, all 46 patients with baseline resistance-associated variants achieved a SVR. Neither of the 2 patients who relapsed after treatment with sofosbuvir-velpatasvir-voxilaprevir had treatment-emergent resistance, whereas both patients who relapsed after receiving sofosbuvir-velpatasvir had the Y93H variant, which is associated with resistance to NS5A inhibitors, at time of relapse.
Safety
None of the 611 patients receiving 8 weeks of sofosbuvir-velpatasvir-voxilaprevir in the POLARIS-2 and POLARIS-3 trials discontinued study treatment owing to adverse events (Table 3). Two patients (<1%) receiving 12 weeks of sofosbuvir-velpatasvir in POLARIS-2 discontinued treatment prematurely owing to adverse events: a 70-year-old white man who discontinued treatment on day 4 owing to an upper respiratory tract infection, and a 54-year-old white woman discontinued after 81 days of dosing as a result of Clostridium difficile colitis. In the POLARIS-3 trial, 1 (1%) patient receiving sofosbuvir-velpatasvir discontinued treatment on day 6 after experiencing a pelvic fracture.
A total of 15 patients (3%) in the sofosbuvir-velpatasvir-voxilaprevir group of POLARIS-2 had serious adverse events; no single patient had more than 1 event and no single event was experienced by more than 1 patient (Supplementary Table 5). In the sofosbuvir-velpatasvir group, 7 patients (2%) had a total of 11 events. The only serious adverse event that occurred in both treatment groups was pyelonephritis. In POLARIS-3, 2 patients (2%) in the sofosbuvir-velpatasvir-voxilaprevir group had a total of 4 serious adverse events, and 3 patients (3%) in the sofosbuvir-velpatasvir group had 1 serious adverse event each (Table 3). No single serious adverse event was experienced by more than 1 patient (Supplementary Table 6).
Overall, 72% of patients receiving sofosbuvir-velpatasvir-voxilaprevir in POLARIS-2 had adverse events, compared with 69% of those receiving sofosbuvir-velpatasvir (Table 3). The most common adverse events in the sofosbuvir-velpatasvir-voxilaprevir group were headache (27% of patients), fatigue (21%), diarrhea (18%), and nausea (16%), compared with headache (23%), fatigue (20%), nausea (7%), and diarrhea (9%) in the sofosbuvir-velpatasvir group. In the POLARIS-3 trial, 75% of patients receiving sofosbuvir-velpatasvir-voxilaprevir experienced adverse events, the most common of which were fatigue (25%), headache (25%), nausea (21%), and diarrhea (15%). Among patients receiving sofosbuvir-velpatasvir, 74% had adverse events, the most common of which were headache (29%), fatigue (28%), nausea (9%), and upper abdominal pain (6%). Among patients receiving 8 weeks of sofosbuvir-velpatasvir-voxilaprevir across trials, 95% of the events of diarrhea or nausea were mild in severity, 5% were grade 2, and none were grades 3 or 4.
Hematologic and chemistry laboratory abnormalities were uncommon in all groups (Table 3 and Supplementary Tables 8 and 9). Hyperglycemia occurred at a higher rate in the POLARIS-3 study compared with the POLARIS-2 study, which may be attributable to the difference in study populations and the association of glucose dysregulation with liver disease. None of the laboratory abnormalities in any treatment group was associated with a clinical adverse event.
|
|
| |
| |
|
|
|