| |
Early antibody therapy can induce long-lasting
immunity to SHIV / Dual Antibody Therapy
|
| |
| |
Download the PDF here
Nature March 3 2017
Monkeys Suppress HIV-Like Virus for Extended Period after Dual-Antibody Treatment - NIAID Press
Announcement:https://www.niaid.nih.gov/news-events/monkeys-suppress-hiv-virus-extended-period-after-dual-antibody-treatment
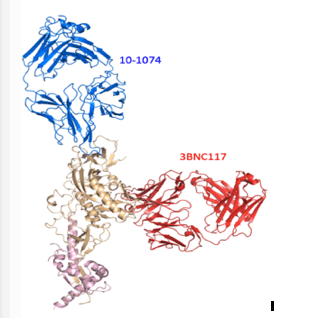
This protein-structure model shows how the broadly neutralizing antibodies 10-1074 (blue) and 3BNC117 (red) that were infused into the monkeys each bind to a different site on the SHIV spike (beige and pink), helping prevent the virus from escaping the antibodies.
Giving monkeys two powerful anti-HIV antibodies immediately after infection with an HIV-like virus enabled the immune systems of some of the animals to control the virus long after the antibodies were gone, scientists at the National Institutes of Health and The Rockefeller University have found.
The researchers inoculated 13 monkeys with simian-human immunodeficiency virus, or SHIV. Three days after inoculation, when an infection clearly was established, the scientists gave each monkey three intravenous infusions of two potent, broadly neutralizing HIV antibodies over a two-week period. These antibodies, called 3BNC117 and 10-1074, each bind to a different site on SHIV, helping to neutralize the virus and facilitating its clearance by the immune system.
The antibody infusions suppressed SHIV to levels near or below the limit of detection by standard assays for as long as six months. Once antibody levels had fallen very low, the virus rebounded in all but one animal. Then, five to 22 months later, the immune systems of six of the monkeys spontaneously regained control of the virus and brought it down to undetectable levels for another five to 13 months. These six controller monkeys had continuously maintained healthy levels of key immune cells after receiving the antibody infusions.
In addition to the six controller animals, four other monkeys that failed to regain complete control of the virus nevertheless maintained extremely low levels of SHIV in the blood and healthy levels of key immune cells for two to three years after infection. Thus, ten of the 13 monkeys benefitted from antibody immunotherapy, according to the researchers.
To determine whether immune cells called CD8+ T cells played a role in extended control of SHIV, the scientists gave the six controller monkeys an antibody that targets and depletes these cells. Infusion of this antibody immediately increased the amount of SHIV in the monkeys' blood as CD8+ T-cell levels decreased. The researchers concluded that CD8+ T cells controlled SHIV replication after therapeutic antibody infusion.
Studies are underway to test whether waiting to administer the therapeutic antibody infusions until two to six weeks after SHIV infection-timing that more closely resembles how soon an HIV-infected person is usually diagnosed-also enables monkeys to control SHIV.
-----------------------------------------
Early antibody therapy can induce long-lasting immunity to SHIV
Nature
Published online 13 March 2017
"As proof of concept, our results demonstrate that a SHIV infection, established during the acute phase, can, in fact, be controlled. They also suggest that a delicate balance may exist between (1) preservation of helper CD4+ T cells, (2) the size and stability of the virus reservoir, and (3) the continuous production of sufficient quantities of antigen to generate a potent and sustained CD8+ T-cell response. Although rhesus macaque infections with SHIV differ from HIV-1 infections in several important ways, immunotherapy should be explored as a way of controlling systemic dissemination of virus, of containing damage to the CD4+ T-cell lineage, and of mobilizing a robust immune response that may be capable of controlling the infection in humans."
Highly potent and broadly neutralizing anti-HIV-1 antibodies (bNAbs) have been used to prevent and treat lentivirus infections in humanized mice, macaques, and humans1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12. In immunotherapy experiments, administration of bNAbs to chronically infected animals transiently suppresses virus replication, which invariably returns to pre-treatment levels and results in progression to clinical disease. Here we show that early administration of bNAbs in a macaque simian/human immunodeficiency virus (SHIV) model is associated with very low levels of persistent viraemia, which leads to the establishment of T-cell immunity and resultant long-term infection control. Animals challenged with SHIVAD8-EO by mucosal or intravenous routes received a single 2-week course of two potent passively transferred bNAbs (3BNC117 and 10-1074 (refs 13, 14)). Viraemia remained undetectable for 56-177 days, depending on bNAb half-life in vivo.
Moreover, in the 13 treated monkeys, plasma virus loads subsequently declined to undetectable levels in 6 controller macaques. Four additional animals maintained their counts of T cells carrying the CD4 antigen (CD4+) and very low levels of viraemia persisted for over 2 years. The frequency of cells carrying replication-competent virus was less than 1 per 106 circulating CD4+ T cells in the six controller macaques. Infusion of a T-cell-depleting anti-CD8β monoclonal antibody to the controller animals led to a specific decline in levels of CD8+ T cells and the rapid reappearance of plasma viraemia. In contrast, macaques treated for 15 weeks with combination anti-retroviral therapy, beginning on day 3 after infection, experienced sustained rebound plasma viraemia when treatment was interrupted. Our results show that passive immunotherapy during acute SHIV infection differs from combination anti-retroviral therapy in that it facilitates the emergence of potent CD8+ T-cell immunity able to durably suppress virus replication.
After an initial and massive burst of HIV replication15, viraemia is incompletely controlled by the emergence of a virus-specific CD8+ T-cell response, leading to a chronic phase during which the infection is never cleared16, 17. Virions that integrate their DNA into the host cell genome become a source of virus production, which persists despite anti-retroviral treatment over extended periods of time and gives rise to a recrudescent progressive infection when treatment is stopped18. This virus reservoir persists for the lifetime of the infected individual19.
Nonetheless, initiation of combination anti-retroviral therapy (cART) within 6 months of HIV-1 infection may limit damage to the immune system as well as the size of the reservoir, as measured by preservation of anti-virus T-cell responses and lower levels of cell-associated viral DNA20. In a rare subpopulation of people with HIV-1 infection, cART initiated during the early phases of infection resulted in sustained control of viraemia to very low levels, after treatment interruption21. Durable control of plasma viraemia has also been reported in some SIVsmE660-infected rhesus macaques after anti-retroviral treatment, started 24 or 72 h after intravenous inoculation, was discontinued22.
Because bNAbs suppress viraemia3, 23, accelerate the clearance of cell-free HIV-1 virions24, enhance clearance of infected cells6, 25, 26, and boost host humoral immunity23 in humans, they have the potential to mitigate deleterious events occurring during the acute infection and possibly alter the long-term clinical course. Consistent with this hypothesis, experiments in humanized mice indicate that early intervention with bNAbs may be more effective in preventing establishment of the reservoir than cART5. However, humanized mice do not have intact immune systems, and fail to sustain infection beyond 3-4 months.
We previously reported that initiation of bNAb monotherapy, 12 weeks after SHIVAD8-EO inoculation, controls plasma viraemia in macaques for 1 or 2 weeks, after which resistant viral variants emerge12. Thus, it is problematical whether even early combination bNAb immunotherapy might durably control viraemia. To address this question, combination 10-1074 (ref. 13) and 3BNC117 (ref. 14) immunotherapy, which targets non-overlapping epitopes on the envelope spike, was administered to rhesus monkeys at the earliest possible time (that is, 3 days after infection) when we were certain that a SHIVAD8-EO infection had been established.
SHIVAD8-EO infection resembles HIV-1 infection in several important ways. It is R5 tropic, generates sustained levels of plasma viraemia in inoculated macaques, exhibits a Tier 2 neutralization sensitivity phenotype, produces resistant variants in bNAb- and ART-treated animals, and causes irreversible depletions of CD4+ T cells in infected monkeys (refs 12 and 27, 28, 29, and Y.N. and M.A.M., unpublished observations). Infection leads to symptomatic immunodeficiency associated with opportunistic infections and a fatal clinical outcome in untreated monkeys.
In the initial experiment to assess the potency of combination bNAbs during acute infection, six macaques were inoculated intrarectally with 1,000 TCID50 (50% tissue culture infective dose) of SHIVAD8-EO, which is sufficient to infect all animals challenged by this route11. Beginning on day 3 after inoculation, each monkey received a single course of three weekly intravenous bNAb infusions (on days 3, 10, and 17 after infection) of 10-1074 (ref. 13) plus 3BNC117 (ref. 14). Compared with untreated animals, extremely low levels of plasma viraemia could be detected in two of the six bNAb recipients during the first 30 days of infection (Fig. 1a). Plasma viraemia in the other four macaques was not measurable (<100 SHIV RNA copies per millilitre) using standard PCR with reverse transcription (RT-PCR) assays.
All six monkeys infused with the bNAbs experienced sustained periods of virus suppression lasting 56-177 days, at which point rebound viraemia occurred in five of the six treated macaques (Extended Data Fig. 1). Plasma viraemia remained below levels of detection in the sixth macaque (DFIK) during the first 150 days of infection/treatment (Extended Data Fig. 1f). As expected, the time to virus rebound was directly related to the concentration of bNAb in the plasma. For example, the SHIVAD8-EO rebound in macaque MVJ occurred on day 56 when the level of 3BNC117 decayed below 1 μg ml-1 in plasma, whereas monkeys DEPH and DEWP experienced prolonged suppression of viraemia before rebound was observed (Extended Data Fig. 1a, d, e). We conclude that a single 2-week course of combination antibody therapy with 3BNC117 and 10-1074, administered early after an intrarectal infection, can control SHIVAD8-EO viraemia for up to 177 days.
To determine whether low-level viral replication before virus rebound persisted despite apparent control of viraemia, we performed ultrasensitive nested RT-PCR (qRT-PCR) on plasma samples from animals with viral loads below levels of detection by standard methods (Extended Data Table 1). For example, in macaque DEPH, which did not generate a detectable virus rebound until day 177 (Extended Data Fig. 2d), the ultrasensitive assay measured fewer than two viral RNA copies per millilitre in plasma, between days 34 and 106 after infection, and ten RNA copies per millilitre on day 141 after infection, a month before rebound. We conclude that low levels of virus are continuously being produced systemically and released into the circulation in the infected and bNAb-treated macaques, even when plasma viraemia is not detected by standard assays.
Two patterns of 'post-rebound' plasma viraemia were observed in the six bNAb-treated monkeys. In the first (DFIK, MVJ, and DEWP; controller (Fig. 2a-c)), the rebound SHIVAD8-EO RNA loads in plasma returned to undetectable levels after a maximum of 20 weeks and remained so for an additional 20-30 weeks of observation. The second pattern occurred in monkeys DEPH, DFKX, and DFFX (non-controller (Fig. 2d-f)). These three macaques never fully controlled their viraemia after rebound and displayed viral loads of 1 x 102 to 103 RNA copies per millilitre between 110 and 130 weeks after virus inoculation.
Sustained control of plasma viraemia by early bNAb therapy after a mucosal SHIVAD8-EO challenge in three out of six monkeys suggested that the same intervention might also be effective against challenge by the intravenous route, for which a smaller virus inoculum is typically required to establish an infection. Macaques were therefore inoculated with either 100 or 1,000 TCID50 of SHIVAD8-EO intravenously and then administered three weekly bNAb infusions starting on day 3 after infection. As shown in Fig. 1b, the four recipients of 1,000 TCID50 of virus showed peak plasma virus loads ranging from 4.2 x 103 to 1.1 x 104 RNA copies per millilitre during the first 2 weeks of bNAb administration. Two of the three recipients (DEMR and DEHW) inoculated with 100 TCID50 of virus intravenously had lower levels of plasma viraemia during the early treatment period, and in the third animal (DEBA), viral loads remained below the level of detection throughout this time (Fig. 1c). Viraemia declined to undetectable levels by 30 days after bNAb administration and remained suppressed for 48-110 days after infection in all seven combination bNAb-treated monkeys inoculated by the intravenous route (Extended Data, Fig. 2). As previously observed in the intrarectally challenged animals, virus rebound in the intravenously inoculated monkeys was directly associated with the decline of circulating bNAb levels.
To determine whether virus rebound was due to declining levels of bNAbs in the plasma or to the emergence of mutation(s) conferring resistance, we sequenced plasma viral RNA from the four monkeys (DELV, DF06, DEWL, and MAF), generating detectable viraemia at a time when the 3BNC117 monoclonal antibody (mAb) was not detectable, but 10-1074 mAb concentrations remained above threshold levels (see Extended Data Fig. 2). When samples collected at the peak of virus rebound from these 4 animals were analysed, no changes were observed in amplicons from macaques DELV and DEWL, but 9 out of 18 amplified sequences from macaque DF06 carried the N332S change and 12 out of 24 amplicons from monkey MAF had the S334N substitution, both of which eliminated the glycan at position 332 of gp120 (Extended Data Fig. 3), the epitope targeted by the 10-1074 mAb. This result indicated that resistance to the 10-1074 mAb probably occurred in these two animals during the time when 10-1074, but not 3BNC117, was detectable in the plasma.
Similar to the macaques challenged by the intrarectal route, passive immunotherapy after intravenous infection also led to sustained virus control after the resolution of rebound viraemia in some of the challenged monkeys. In three macaques (DEWL, MAF, and DEMR; controllers), rebound viraemia remained detectable for 42-90 weeks, and was then followed by long periods (25-56 additional weeks) during which virus replication was stably suppressed below the limits of detection (Fig. 3a-c). CD4+ T-cell counts were maintained at a level of 800 cells per microlitre or higher in these three controller animals. In the second group of intravenously challenged animals (monkeys DF06, DELV, DEHW, and DEBA; non-controllers), virus replication was never completely suppressed after rebound (Fig. 3d-g). Failure to control viraemia was associated with CD4+ T-cell loss and progression to AIDS in one (macaque DF06) of these four monkeys.
Humoral and cellular immune responses were assessed before and after SHIVAD8-EO rebound to determine whether anti-viral immunity was associated with controller status. No measurable correlation was observed between virus control and antibody levels. The controllers generated very low levels of anti-gp120 binding antibodies that were barely detectable by enzyme-linked immunosorbent assay. Anti-viral CD8+ T-cell responses were measured at four different times during the bNAb treatment, virus rebound, and post-rebound phases of their infections in both controllers and non-controllers. Although anti-SIVmac239 Gag CD8+ T-cell responses were higher in animals challenged by the intravenous route, no major differences were observed between controllers and non-controllers (Extended Data Fig. 4a). Furthermore, the polyfunctionality of the CD8+ T-cell responses and distribution of CD8+ T-cell memory subsets in intravenously inoculated controllers and non-controllers at different times after infection, treatment, and rebound were comparable (Extended Data Fig. 4b, c).
To determine whether CD8+ T cells might, in fact, be mediating the sustained suppression of virus replication, we initially administered the CD8 T-cell-depleting mAb MT807R1, which is specific for the CD8α chain, to all six controller macaques (MVJ (at day 434 after infection), DEWP (at day 434 after infection), DFIK (at day 336 after infection), DEMR (at day 609 after infection), MAF (at day 917 after infection), and DEWL (at day 917 after infection)) (Fig. 4a-f). All animals responded with a burst of plasma viraemia, reaching levels between 105 and 107 viral RNA copies per millilitre, which subsequently declined to baseline in all of the monkeys, except for DFIK.
Before the administration of the anti-CD8α mAb, quantitative virus outgrowth assays were performed on samples collected from the six controllers to measure the frequency of circulating CD4+ T cells releasing replication-competent virions. As shown in Extended Data Table 2, fewer than 1 in 106 CD4+ T cells carrying infectious virus was detected in all of the controller monkeys immediately before infusion of the depleting anti-CD8α mAb. Interestingly, in five of the six anti-CD8α mAb recipients in which plasma viraemia had returned to baseline levels after mAb infusion, the frequency of cells carrying replication-competent virus, determined by virus outgrowth assays, was also less than 1 in 106 CD4+ T cells (Extended Data Table 2).
A major deficiency of using the anti-CD8α mAb to deplete CD8+ T cells is that NK, NKT, and γσ T cells are also targeted for depletion, as documented by fluorescence-activated cell sorting (FACS) analyses for monkeys MVJ, DEWL, and DEMR (Extended Data Fig. 5a, b). We therefore administered the CD8b255R1 anti-CD8β mAb, which specifically targets macaque CD8+ T cells, to deplete CD8+ T cells in these same three controller monkeys. As shown in Fig. 4a-c (red arrows), infusion of the anti-CD8β mAb caused an immediate increase in plasma virus loads, a decline in levels of CD3+CD8+ T cells (Extended Data Fig. 5c), and no changes in circulating CD3-CD8+ cells (Extended Data Fig. 5d). We conclude that CD8+ T cells are responsible for control of virus replication in controller macaques and depletion of this subset leads to recrudescence of viraemia.
Although the non-controller monkeys failed to suppress plasma viraemia to undetectable levels, four (DFKX, DFFX, DEHW, and DEBA) of seven of these macaques maintained low virus loads (105-385 RNA copies per millilitre) and did not experience significant changes in levels of circulating CD4+ T cells for 2-3 years after SHIVAD8-EO infection (Figs 2 and 3). Taken together, this indicates that 10 (6 controllers and 4 non-controllers) of the 13 bNAb-treated monkeys benefited from early immunotherapy.
As a control for bNAb immunotherapy during the acute SHIVAD8-EO infection, three macaques were inoculated by the intrarectal route and treated daily with cART for 15 weeks, starting on day 3 after infection. This course of cART corresponded to the mean period during which 2 weeks of bNAb therapy suppressed virus replication before rebound. cART therapy controlled and maintained undetectable levels of viraemia for the entire 15-week treatment period (Extended Data Fig. 6). However, all three animals developed high sustained levels of plasma viraemia after cessation of cART and none became controllers.
We speculate that the continuous production of low levels of progeny virions, as measured by ultrasensitive RT-PCR (Extended Data Table 1) during the 50- to 140-day period before virus rebound in the antibody treated macaques, could drive the formation of immune complexes. Antigen-presenting dendritic cells expressing activating Fc receptors can bind to these immune complexes, leading to their activation and efficient antigen processing for presentation and cross-presentation to CD4+ and CD8+ T cells30. In contrast, the near-complete inhibition of virus replication by the cART regimen used may limit the amount of viral antigen available to induce immunity.
As proof of concept, our results demonstrate that a SHIV infection, established during the acute phase, can, in fact, be controlled. They also suggest that a delicate balance may exist between (1) preservation of helper CD4+ T cells, (2) the size and stability of the virus reservoir, and (3) the continuous production of sufficient quantities of antigen to generate a potent and sustained CD8+ T-cell response. Although rhesus macaque infections with SHIV differ from HIV-1 infections in several important ways, immunotherapy should be explored as a way of controlling systemic dissemination of virus, of containing damage to the CD4+ T-cell lineage, and of mobilizing a robust immune response that may be capable of controlling the infection in humans.
|
|
| |
| |
|
|
|