| |
Of Mice, Macaques, and Men: Broadly Neutralizing Antibody Immunotherapy for HIV-1 - Review
|
| |
| |
Cell Host Microbe Aug 2017 - Yoshiaki Nishimura1 and Malcolm A. Martin1,*
1Laboratory of Molecular Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, BethesdaMD20892
Download the PDF here
The neutralizing antibodies targeting the HIV-1 envelope protein have been a major focus for HIV therapy. Early studies with anti-HIV-1 neutralizing monoclonal antibodies (mAbs) administered to infected individuals showed some promise, as they resulted in transient reductions in plasma viremia in some recipients. However, resistant viral variants rapidly emerged. A major development during the past 6 to 7 years has been the isolation and characterization of highly potent and broadly neutralizing mAbs (bNAbs) from infected individuals known as "elite neutralizers." These "next-generation" bNAbs have been tested in animal model systems and shown to effectively control virus replication, particularly following combination immunotherapy. The success of these preclinical animal studies has led to human clinical trials using an individual bNAb for therapy. This review examines recent findings from animal models and human clinical trials and discusses the future use of bNAbs for HIV-1 treatment.
Main Text
Introduction
One of the most significant accomplishments during the 35 years of the HIV-1 AIDS epidemic has been the discovery, development, and use of combination anti-retroviral therapy (cART) to treat virus-infected individuals. More than 40 FDA-approved antiretroviral drugs and drug combinations in seven different pharmaceutical classes are currently available. This breakthrough has resulted in extraordinary reductions in plasma virus loads, a decrease in AIDS-related mortality by more than 80% in the developed world, and lower rates of HIV-1 transmission, particularly in areas where the virus is endemic (Samji et al., 2013, Wada et al., 2013, Walensky et al., 2006). Although disease progression has been arrested, life expectancy has been increased, and the quality of life of an infected person has been dramatically improved, these benefits require daily, lifelong therapy and are accompanied by numerous side effects. Furthermore, and despite its impressive effectiveness, cART fails to eradicate the virus in an infected individual primarily because these drugs are unable to eliminate the latent virus reservoir (Finzi et al., 1997, Wong et al., 1997). As a result, when or if cART is discontinued, the suppressed virus rapidly rebounds, usually reaching pre-treatment levels of viremia (Davey et al., 1999).
Prior to the development of effective cART, anti-HIV-1 neutralizing antibodies were considered as a possible therapy for virus-infected individuals. The first approaches, in the late 1980s, employed infusions of serum from patients possessing high levels of antibodies recognizing the immunodominant p24 (or capsid) region of the structural Gag protein (Jackson et al., 1988, Karpas et al., 1990). When it became firmly established in the 1990s that the HIV-1 envelope protein, not the capsid protein, was the only target of neutralizing antibodies (NAbs), the first generation of neutralizing anti-HIV-1 monoclonal antibodies (mAbs) were isolated and characterized; they were used to treated infected individuals in the early 2000s. These antibodies recognized various portions of the viral envelope, which is comprised of the glycoproteins gp41 and gp120, that facilitate virus entry by engaging the CD4 receptor and CCR5 or CXCR4 co-receptors. The envelope poses challenges to sustained antibody recognition due not only to heavy glycosylation, but also to variable loops (V1-5) that can change to escape immune pressures.
Among the first NAbs that were shown to target the viral envelope included 2G12 (targeting gp120 N-linked glycans), 2F5 and 4E10 (both binding to epitopes in the gp41 ectodomain). Although these mAbs were safe and well tolerated, they only modestly suppressed viremia in some recipients, who rapidly developed resistance to the 2G12 component of the administered antibody cocktail (Jaworski et al., 2017, Stephenson and Barouch, 2016). The subsequent development of high through-put neutralization assays and standardized panels of HIV-1 pseudotyped viruses, the identification of a small subset of HIV-1-infected individuals, designated elite neutralizers, who generated high titers of broadly neutralizing antibodies (bNAbs), and the development of single B cell cloning technology, all facilitated the discovery of "new generation" mAbs that have recently been used for prevention and treatment studies (Bonsignori et al., 2017, Burton and Mascola, 2015, Escolano et al., 2017, McCoy and Burton, 2017). A summary of immunotherapeutic interventions using these bNAbs to suppress infections of humanized mice, macaques and humans is the subject of this review.
Preclinical animal studies evaluating HIV-1 NAbs in vivo have utilized two systems: 1) SIV/HIV chimeric viruses (SHIVs) (Shibata et al., 1991) and macaque monkeys for nearly two decades; and 2) HIV-1 and humanized mice (hu-mice) (Brehm et al., 2010, Traggiai et al., 2004) for the past 5 years. Although no perfect animal model for human disease exists, each of these systems has advantages and disadvantages for assessing new NAbs that target the HIV-1 envelope glycoprotein (Table 1).
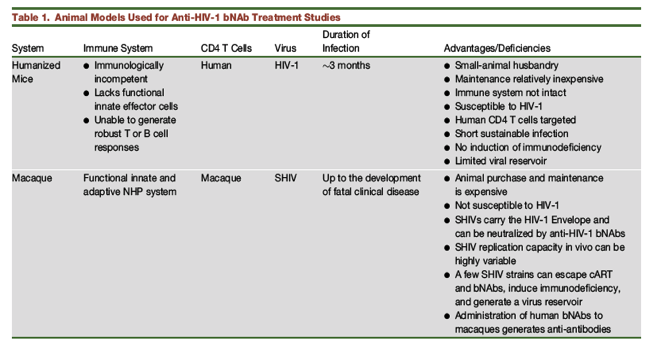
The most compelling reason for using hu-mice to assess bNAb effectiveness for treatment is that the engrafted human CD4+ T cells are fully susceptible to the inoculated virus, HIV-1. However, because these natural HIV-1 cell targets are irreversibly depleted and not replenished in this model, a chronic virus infection is only sustainable for 3 to 4 months, thereby precluding assessments of important ancillary issues such as the establishment and maintenance of a virus reservoir in resting human memory CD4+ T lymphocytes. Furthermore, the mouse immune system in not intact in hu-mice, and inoculated animals are unable to mount robust T- or B-cell responses. These animals also fail to develop HIV-1-induced immunodeficiency.
The principal advantage of the SHIV macaque model is that the in vivo infection can be studied from the time of virus acquisition to death from immunodeficiency. This is a model with an intact immune system capable of generating adaptive T- and B-cell responses as well as the development of a latent virus reservoir. There are, however, several downsides to this expensive, large-animal model. First, although the immune system is intact, macaque CD4+ T cell targets are resistant to HIV-1; this requires the use of SHIVs, which are SIVs that carry an HIV-1 envelope and have been adapted to replicate efficiently in monkey CD4+ T lymphocytes as a consequence of repeated serial passaging in rhesus macaques. An intrinsic property of HIV-1 is its replicative vigor and genetic variability in vivo, both of which confer the capacity to escape immunologic and pharmacologic pressure. Unfortunately, many currently available SHIV challenge viruses do not possess this property and are unable to resist immunologic or cART pressure. This can lead to long-term and irreversible suppression of virus replication rather than robust and sustained production of progeny virions and unrelenting damage to the immune system, a signature of HIV-1 infections in humans. Finally, the infusion of neutralizing human mAbs into monkeys is highly immunogenic and rapidly elicits anti-antibodies, which prematurely eliminate and diminish the effectiveness of bNAbs in vivo (Gautam et al., 2016).
In the immunotherapy experiments described below, members of three different bNAb classes have been used to treat infected hu-mice, macaques, or human patients. Antibodies targeting the HIV-1 CD4 binding site of the envelope glycoprotein included b12 (Burton et al., 1994), VRC01 (Zhou et al., 2010), 3BNC117 (Scheid et al., 2011), VRC07-523 (Rudicell et al., 2014), and 45-46G54W (Diskin et al., 2011). bNAbs binding to the high-mannose patch downstream of the gp120 V3 loop included PGT121 (Walker et al., 2011), PGT128 (Walker et al., 2011), and10-1074 (Mouquet et al., 2012). PG16 (Walker et al., 2009), used only in hu-mice, targets the N160 glycan and basic residues in the gp120 V2 loop.
HIV-1 Hu-mouse Model
Evaluations of the potency of next generation bNAbs as therapy for virus-infected animals began with studies using the hu-mouse model (Table 2). In this system, antibodies were usually administered 2 to 3 weeks following virus inoculation, coinciding with, or immediately following, the initial burst of virus replication in vivo. Klein et al. (2012)) reported that bNAb monotherapy of hu-mice led to a transient decrease (0.23 to 1.5 log10) of plasma viremia and the rapid emergence of antibody-resistant HIV-1 variants. On the other hand, a combination of five bNAbs (3BC176, PG16, 45-46G54W, PGT128, and 10-1074, and designated pentamix) suppressed virus replication for an average of 60 days in seven of eight treated animals, and no evidence of resistance was observed in the rebounding virus population. Focusing on HIV-1-infected hu-mouse recipients of the PG16, 3BNC117, and 10-1074 mAb trimix, Halper-Stromberg et al. (2014)) reported 1) that virus rebound was significantly delayed (74 to 107 days post-infection [PI]) in 10 of the 18 animals administered the trimix compared to mice treated for 40 days with cART alone (28 to 84 days PI); and 2) that a bNAb trimix carrying the Fc domain mutation GRLR, which prevents binding to human and mouse Fc-receptors, controlled virus replication in vivo for a shorter period of time than the WT bNAb trimix.
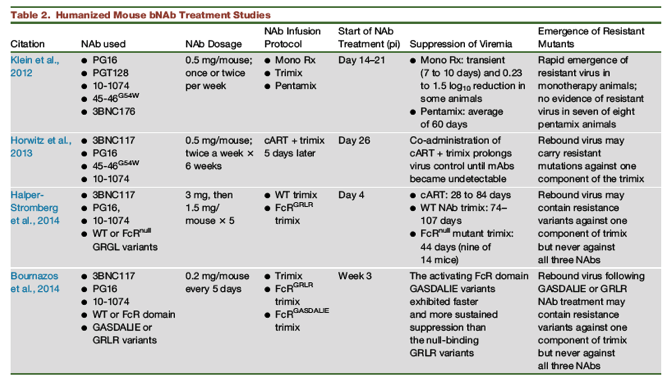
To examine how cART might influence bNAb effectiveness in vivo, Horwitz et al. (2013)) treated hu-mice, infected with an HIV-1 strain carrying a CCR5 tropic envelope (YU-2), with cART for 3 weeks and then added biweekly bNAb trimix (45-46G54W, PG16, and 10-1074 mAbs) infusions 5 days after starting cART. The trimix immunotherapy was continued for 4 additional weeks following cART termination (Horwitz et al., 2013). Viremia was suppressed to below the limits of detection at day 30 post-cessation of trimix administration and rebounded when plasma mAb levels became unmeasurable. Nucleotide sequencing the gp120 clones from samples of rebounding virus revealed the presence of different mutations known to confer resistance to any one of the three components of the trimix; no clones were identified bearing resistance to all three mAbs.
To further assess the role of Fc effector function, Bournazos et al. tested bNAb Fc domain variants bearing either activating (GASDALIE) or non-binding (GRLR) mutations (Bournazos et al., 2014). Recipients of the GASDALIE trimix exhibited faster and more durable control of plasma viremia than hu-mice administered the GRLR trimix, indicating that Fc effector function is an important contributor to bNAb activity in this in vivo model with human CD4+ T cells and HIV-1 components.
SHIV Macaque Model
Studies assessing next generation bNAb control of virus replication in SHIV-infected macaques began approximately one year after the initiation of similar hu-mouse experiments (Table 3). As noted earlier and in Table 1, one of the shortcomings of the macaque model is that some SHIV challenge virus stocks do not exhibit robust replication properties in vivo. These SHIV preparations may be unable to 1) sustain detectable levels of progeny virus production following the emergence of cytotoxic T cell (CTL) responses, which controls the acute infection; 2) generate variants able to resist cART or potent bNAb treatments; and 3) cause irreversible depletions of CD4+ T cells resulting in the development of symptomatic immunodeficiency. These are clinical features that a surrogate model for HIV-1 should possess.ND: Not done
One of the initial experiments evaluating bNAb therapy in chronically infected rhesus macaques utilized the originally described CCR5-tropic and commonly used SHIV-SF162P3 and the bNAbs 3BNC117 and/or PGT121, administered 9 months PI (Table 3; Barouch et al., 2013). Because the SHIV-SF162P3 stock used in this study was resistant to the 3BNC117 mAb, the mono- and combination-immunotherapy regimens employing this bNAb devolved into PGT121-only treatment experiments. Nonetheless, PGT121 therapy of 18 SHIV-SF162P3-infected animals suppressed virus replication for 35 to >100 days and controlled viremia to undetectable levels in three of these monkeys with no virus rebound. Cell-associated viral DNA was reduced 4- to 10-fold in PBMCs, lymph nodes, and GI tract mucosa. Neutralization escape SHIV variants were never detected in the 18 PGT121 treated animals.
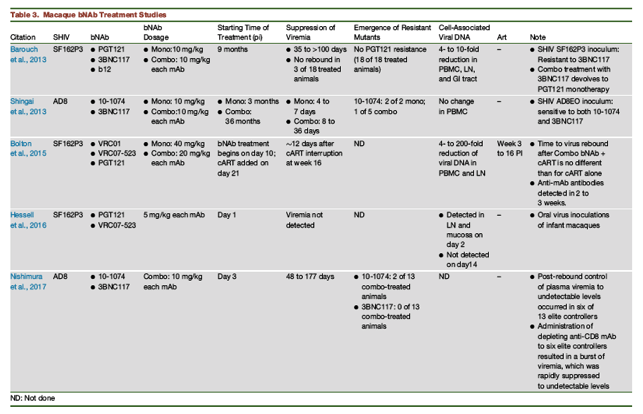
A contemporaneous report evaluated chronically SHIV-AD8-infected monkeys as well as 10-1074 and 3BNC117 mAbs administered individually or in combination at 3 months or 36 months PI, respectively (Shingai et al., 2013). SHIV-AD8 is CXCR5-tropic, generates sustained levels of plasma viremia in inoculated macaques, exhibits a moderate (or Tier 2) neutralization sensitivity phenotype, produces resistant variants in bNAb- and cART-treated animals, and causes irreversible depletions of CD4+ T cells in infected monkeys (Gautam et al., 2012, Nishimura et al., 2010, Shingai et al., 2012, Shingai et al., 2013). Infection usually leads to symptomatic immunodeficiency associated with opportunistic infections and a fatal clinical outcome in untreated monkeys. Shingai et al., (2013)) reported that suppression of viremia following "single-administration" bNAb monotherapy was short and transient (4 to 7 days); 10-1074 mAb-resistant variants rapidly emerged in 2 of 2 of treated macaques (Table 3). Single-infusion combination bNAb treatment at 36 months PI was more effective; virus rebound occurred in asymptomatic animals 18 to 36 days following the initiation of immunotherapy and 10-1074 mAb-resistant virus was detected in one of the five treated monkeys. No change in PBMC-associated viral DNA was measurable.
Bolton et al. (2015)) subsequently examined the effect of adding a 13-week cART regimen to SHIV-SF162P3 infected macaques, previously administered a single infusion of combination VRC07-523 plus PGT121 mAbs on day 10 PI (Table 3; Bolton et al., 2015). Suppression of viremia lasted ∼12 days following cART cessation, which was not different from animals treated with cART alone.
Although most investigations of cART initiation during the first year of infection have reported unrelenting virus replication following treatment interruption, a few studies have suggested that starting cART during this phase of an HIV-1 infection may mitigate irreversible injury to the immune system and possibly limit the size of the latent virus reservoir (Buzon et al., 2014, Jain et al., 2013). Durable control of HIV-1 and SIV replication after stopping cART has also been previously reported when cART was initiated during very early phases of these infections (Lifson et al., 2000, Sáez-Cirión et al., 2013). Furthermore, a 1996 study reported that the transfer of immunoglobulin purified from an SIV-infected, long-term non-progressor, on days 1 and 14 after SIVmacE660 infection, resulted in sustained control of plasma viremia (< 103 RNA copies/mL) in four of six infused animals (Haigwood et al., 1996). SIVmacE660 is an uncloned SIV stock that is less sensitive to neutralizing antibodies than the more commonly used SIVmac239 and SIVmac251 challenge viruses (Goldstein et al., 1994).
Two recent studies have been conducted assessing combination bNAb treatment during acute SHIV infections of macaques. In the first, 1-month-old rhesus monkeys were orally inoculated with SHIV-SF162P3 and treated with a combination of PGT121 and VRC07-523 mAbs on day 1 PI (Hessell et al., 2016). Pairs of animals were euthanized at various times during the first 2 weeks of infection, and levels of viral RNA or DNA were measured in the blood and tissues of treated and untreated monkeys. In bNAb recipients, low levels of SHIV DNA were detectable in mucosa and lymph nodes proximal and distal to the oral exposure site 24 hr following combination mAb administration (viz. on day 2 PI). However, no virus was measurable in tissues of the treated macaques examined on day 14 PI, and plasma viremia was never detected at any time point sampled. These results indicate that virus acquisition occurred as rapidly as 24 hr PI in the 1-month-old newborn rhesus model.
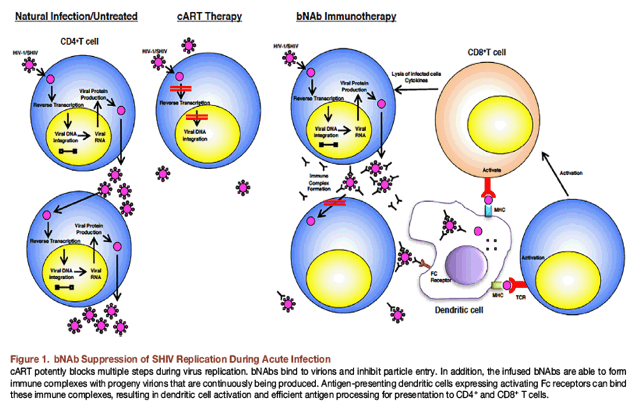
In the second study evaluating bNAb therapy during the acute infection, 13 macaques were inoculated with SHIV-AD8 by intrarectal or intravenous routes and administered 10-1074 plus 3BNC117 mAbs on days 3, 10, and 17 PI (Nishimura et al., 2017). In this infected animal cohort, combination bNAb therapy controlled virus replication for 48 to 177 days, at which time rebound viremia occurred. Unexpectedly, the rebounding virus subsequently declined to undetectable levels over a 20- to 90-week period in six of the 13 bNAb-treated monkeys. Quantitative virus outgrowth assays revealed that the frequency of circulating CD4+ T cells in these controller monkeys, which were capable of releasing infectious virus, was exceedingly low (<1 in 106 cells). No measurable correlation was observed between virus suppression and humoral or cell-mediated immune responses in these elite controllers. However, administration of depleting anti-CD8 mAb to these animals resulted in a burst of viremia, which rapidly declined to undetectable levels when CD8+ T cells reappeared. In the group of "non-controller" bNAb-treated macaques, four of seven maintained low levels (105 to 385 RNA copies/mL) of viremia at 2 to 3 years PI. These results indicate that long-lasting elite controller status can be conferred by administering combination bNAbs very early during the acute SHIV-AD8 macaque infection. This bNAb-mediated sustained suppression of virus replication did not occur following daily cART administration between days 3 and 105 PI; this very likely reflects a delicate balance between: 1) the preservation of helper CD4+ T cells during the acute infection; 2) the presence of high-affinity NAbs for sufficient periods of time to generate immune complexes capable of stimulating a potent and sustained CD8+ T cell response; and 3) the size and stability of the virus reservoir (Figure 1). In this regard, these findings are consistent with earlier studies reporting the administration of polyclonal, neutralizing anti-SIV immunoglobulin to rhesus monkeys 7 days post-challenge with the molecularly cloned and highly pathogenic SIVmac239. This treatment elicited an unusually potent CD8+ T cell response, which suppressed plasma viremia for up to 2 years in four of the six treated animals (Iseda et al., 2016, Yamamoto et al., 2009).
bNAb Treatment of HIV-1-Infected Patients
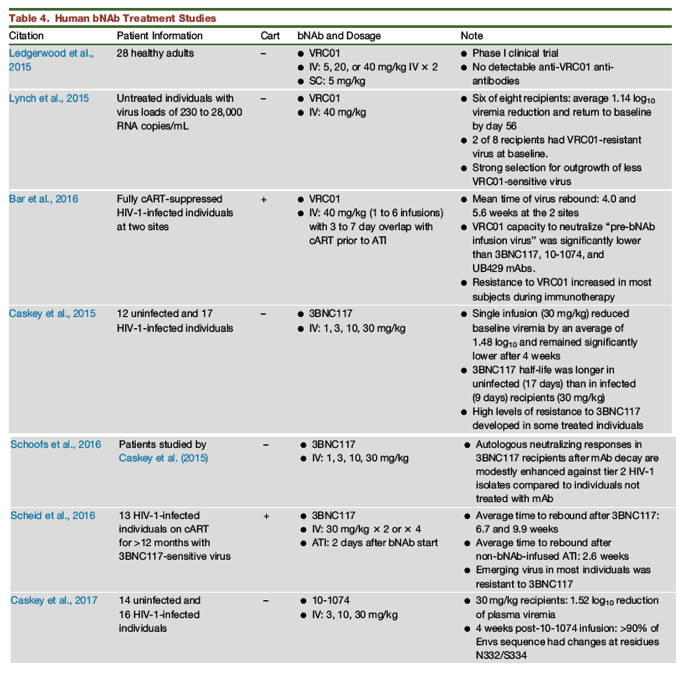
Human clinical bNAb trials have been conducted for the past 2 years (Table 4). Thus far, only monotherapy regimens have been administered to chronically HIV-1-infected patients and, in most instances, have recapitulated some of the findings obtained in the hu-mouse and SHIV macaque models. When observed, virus suppression was invariably transient and seen at the highest bNAb doses administered (30 to 40 mg/kg). Outgrowth of less sensitive and/or resistant HIV-1 strains was frequently seen in the rebounding virus populations.
In one of the early studies, Ledgerwood et al. (2015)) reported that the terminal half-life of VRC01 was 14 days in 28 uninfected healthy adults, and no antibodies directed against the infused VRC01 were detected (Ledgerwood et al., 2015). Lynch et al. (2015)) administered VRC01 to untreated HIV-1-infected individuals and reported an average 1.14 log10 reduction of plasma viremia in six of eight treated patients, with a return of virus loads to baseline levels by day 56 (Lynch et al., 2015). Two of the eight bNAb recipients harbored VRC01-resistant HIV-1 variants prior to treatment, and immunotherapy in these individuals was ineffective. Outgrowth of HIV-1 carrying gp120 mutations blocking VRC01 contact sites was identified in rebound virus samples.
In a phase 1 VRC01 clinical trial conducted at 2 different sites (University of Pennsylvania and NIAID), fully cART-suppressed, chronically HIV-1-infected individuals received one to six infusions of the mAb (40 mg/kg), which overlapped the interruption of cART by 3 to 7 days (Bar et al., 2016). The mean times to virus rebound were 4.0 and 5.6 weeks at the two sites. The measured VRC01 concentration at the time of virus rebound in 23 of 24 of the bNAb recipients was >50 μg/mL of plasma. Although mAb treatment slightly delayed virus rebound compared to historical cART interruption controls, suppression of virus replication was not observed beyond 8 weeks. The virus recovered from most subjects exhibited increased resistance to VRC01.
The gp120 CD4 binding-site-targeting 3BNC117 mAb was the first bNAb tested in humans and was administered to both uninfected and virus-infected individuals (Caskey et al., 2015). At a dose of 30 mg/kg, its half-life was longer in uninfected (17 days) than in HIV-1-infected (9 days) patients. A single infusion of 3BNC117 reduced plasma virus loads by an average of 1.48 log10 RNA copies/mL; this suppression of viremia was significant for days 4 to 28 following administration. Using paired pre- and post-treatment samples, levels of resistance to 3BNC117 were observed in some individuals treated with different doses of the mAb. In a followup to this trial, Schoofs et al. (2016)) reported that autologous anti-HIV-1 NAb activity measured in 3BNC117 mAb recipients after bNAb decay was modestly enhanced against a panel of tier 2 HIV-1 isolates, which are moderately sensitivity to antibody neutralization, compared to infected individuals not treated with 3BNC117 (Schoofs et al., 2016). In a subsequent study, Scheid et al. (2016)) administered the 3BNC117 mAb to 13 HIV-1-infected patients who had been 1) continuously treated with cART for more than 12 months and 2) prescreened and shown to be infected with 3BNC117-sensitive virus (Scheid et al., 2016). Compared to a historical cART interruption control averaging 2.6 weeks until HIV-1 rebound, two or four infusions of 3BNC117 (30 mg/kg) resulted in suppression of virus replication for 6.7 or 9.9 weeks, respectively. The virus recovered from most of these bNAb recipients exhibited increased resistance to the 3BNC117 mAb.
In the only clinical assessment of a bNAb that targeted the gp120 high mannose patch C-terminal to the V3 loop, the 10-1074 mAb was administered to both uninfected and infected individuals in a dose-escalation study (Caskey et al., 2017). Eleven of 13 HIV-1-infected recipients of this bNAb (30 mg/kg) had an average 1.52 log10 reduction of viremia; this nadir was reached after an average of 10.3 days following treatment initiation. The two non-responders carried pre-treatment 10-1074-resistant HIV-1 strains. More than 90% of the envelope sequences in plasma collected 4 weeks post-bNAb infusion from the infected individuals responding to the 10-1074 mAb had mutations affecting the targeted gp120 N332 glycan.
Conclusions and Future Directions
With only a few exceptions, bNAb monotherapy suppression of viremia in chronically infected hu-mice, macaques and humans was 1) invariably of short duration, 2) followed by a rebound viremia that reached pre-treatment levels, and 3) associated with the emergence of virus populations that were resistant to the administered neutralizing mAb. Depending on the system, control of virus replication lasted several days (hu-mouse and macaque) or a few weeks (human). The exception was the PGT121-treated SHIV162P3-infected monkeys, in which virus suppression lasted 35 to >100 days (including three of 18 animals that experienced no SHIV rebound) and no bNAb-resistant viral variants ever appeared. The administration of bNAb monotherapy to fully cART-suppressed, HIV-1-infected individuals extended the period of virus control from 4 to 10 weeks (Bar et al., 2016, Scheid et al., 2016), but the rebound virus that emerged in most patients was neutralization resistant.
Combination bNAb treatment has only been conducted in the hu-mouse and macaque models and in the absence of cART. In both systems, the duration of virus suppression was longer than that observed with bNAb monotherapy, and the frequency of emerging resistant virus was low. The development of resistance to one component of an administered bNAb mixture has, in fact, been observed in both animal model systems. Furthermore, it is possible that one bNAb will have a longer half-life than others in the mAb cocktail. In both situations, combination immunotherapy would devolve into monotherapy and resistance to the remaining mAb is likely to occur.
The use of animal models to assess anti-HIV-1 bNAb therapeutic efficacy has both facilitated and accelerated the evaluation of these antibodies in infected humans. In the macaque system, the development of additional SHIVs, bearing envelopes from HIV-1 Clade A and Clade C isolates and possessing a robust replication phenotype in rhesus monkeys, is needed to evaluate treatment strategies to control such strains from the developing world. In addition, the contribution of bNAb Fc-mediated effects on virus suppression has, thus far, only been assessed in the hu-mouse system. Further structure and function characterization of the monkey Fc domain and its interaction with the macaque Fc receptor is needed to design bNAbs better able to clear virus-infected cells in this model.
The history of anti-retroviral drug development has taught that combination cART regimens are required to maintain durable HIV-1 suppression. This will be especially important for bNAb therapy, which must target an HIV-1 swarm carrying a plethora of different envelope proteins, any one of which might be resistant to the mAb administered. It should be noted that even in some bNAb monotherapy trials, patients were prescreened to eliminate carriers of resistant HIV-1 strains (Scheid et al., 2016). It is also known that some bNAbs (e.g., 10E8) are extremely broad and able to neutralize >95% of HIV-1 isolates (Huang et al., 2012), while others exhibit narrower breadth (e.g., 88%). Subtype B virus strains are sensitive to bNAbs targeting the high mannose patch adjacent to gp120 V3, whereas AE subgroup viruses are resistant to this class of mAbs (Caskey et al., 2017, Huang et al., 2014). Clearly, the isolation and characterization of additional potent bNAbs binding to different HIV-1 envelope epitopes and formulated to allow efficient subcutaneous administration will improve the efficacy and expand the use of future immunotherapy regimens.
Although it is not presently known whether combination bNAb treatment of chronically HIV-1-infected individuals will affect the size or functionality of the latent virus reservoir, administration of these mAbs could have other beneficial effects. First, unlike their use in macaques, in which anti-antibodies are consistently induced, bNAbs should be minimally immunogenic in humans; therefore, their activity will be more durable. A single dose of bNAb derivatives with Fc domain changes conferring extended half-lives could provide anti-viral activity lasting several months, compared to currently available cART regimens, which must be administered daily (Ko et al., 2014). In this regard, long-acting, injectable cART formulations are being developed to improve adherence during treatment and for pre-exposure prophylaxis (Boffito et al., 2014).
The results of bNAb treatment in chronically HIV-1-infected individuals and during the acute SHIV infection of macaques are summarized in Figure 2. Compared to untreated subjects, bNAb monotherapy during the chronic virus infection transiently reduces plasma viremia to variable extents and periods of time. It is anticipated, based on hu-mouse and SHIV macaque experiments, that combination bNAb therapy in HIV-1-infected and cART-suppressed patients will be more effective than bNAb monotherapy and that the availability of newly engineered derivatives will greatly extend the period of virus suppression following each dose of mAb.
The role of bNAb treatment during the early phase of primate lentivirus infections is currently still under investigation. In the macaque model, immunotherapy alone at day 3 PI was sufficient to confer elite controller status to 50% of treated animals (Nishimura et al., 2017). The critical question is whether this early treatment regimen will be effective when initiated during later phases of the acute infection. As noted earlier, the success of bNAb intervention on day 3 post-SHIVAD8 inoculation of macaques very likely depends on preventing irreversible injury to the immune system, which accompanies the acute primate lentivirus infection, as well as controlling the size and stability of the latent virus reservoir. The induction of a potent CTL response during the acute infection by bNAb immunotherapy, when CD4+ T lymphocyte function is largely intact, was sufficient to control rebound plasma viremia after bNAb levels in plasma had declined to undetectable levels and to confer elite controller status in controller animals. At present, the duration of this window of bNAb opportunity following the virus inoculation is unknown.
When translated to HIV-1 infection of humans, the time interval from virus exposure to the onset of symptoms to the seeking of medical attention could be 2 to 12 weeks or longer. The success of early bNAb therapy will therefore depend on treatment initiation beginning as early as possible to mitigate permanent damage to the immune system. Ultimately, integrated cART and combination bNAb treatments will have to be designed to maximize the number of HIV-1-infected individuals who achieve elite controller status.
|
|
| |
| |
|
|
|