| |
Brain Damage - Cannabis / Nicotine / Stimulants (Cocaine/Meth), HIV
|
| |
| |
Download the PDF here
"Most stimulants dysregulate the BBB through alterations in tight junction complexes or through oxidative stress and neuroinflammation. Neuroinflammation plays a particularly important role as it contributes to a feed-forward process between BBB dysfunction and neuroinflammation which likely results in the CNS being more vulnerable to infiltration of pathological mediators from the periphery. This is especially important in co-morbid situations of stimulant abuse and immune disorders, like HIV/AIDS. Stimulants not only increase the migration of peripheral HIV-infected leukocytes into the CNS, but once inside the brain, can accelerate the progression to neuroAIDS."
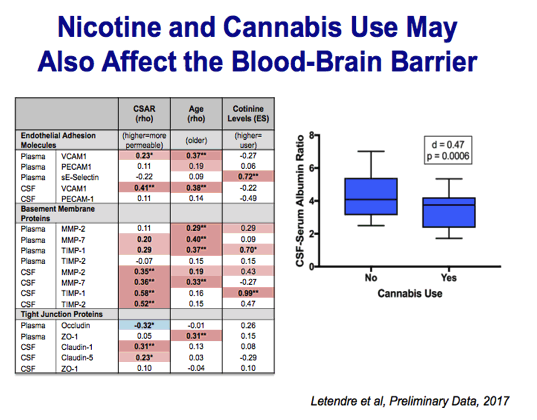
cannabis extracts are known to cause a significant increase in BBB permeability (Agrawal et al. 1989).....The unique smell of cannabis does not arise from cannabinoids, but from over 100 terpenoid compounds (Turner et al. 1980).....http://www.sa.i-pdf.info/h-medicine/142076-2-summary-central-tenet-underlying-the-use-botanical-remedies-that-he.php
"psychostimulants including methamphetamine, MDMA, cocaine, and nicotine, produce BBB dysfunction through alterations in tight junction protein expression and conformation, increased glial activation, increased enzyme activation related to BBB cytoskeleton remodeling, and induction of neuroinflammatory pathways. These detrimental changes lead to increased permeability of the BBB and subsequent vulnerability of the brain to peripheral toxins. In fact, abuse of these psychostimulants, notably methamphetamine and cocaine, has been shown to increase the invasion of peripheral bacteria and viruses into the brain."
"Stimulants not only increase the migration of peripheral HIV-infected leukocytes into the CNS, but once inside the brain, can accelerate the progression to neuroAIDS.
Most stimulants dysregulate the BBB through alterations in tight junction complexes or through oxidative stress and neuroinflammation. Neuroinflammation plays a particularly important role as it contributes to a feed-forward process between BBB dysfunction and neuroinflammation which likely results in the CNS being more vulnerable to infiltration of pathological mediators from the periphery. This is especially important in co-morbid situations of stimulant abuse and immune disorders, like HIV/AIDS.”
-----------------------------
The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation
Sharanya M. Kousik1,2*, T. Celeste Napier1,2 and Paul M. Carvey1,2,3
⋅1 Department of Pharmacology, Rush University Medical Center, Chicago, IL, USA
⋅2 Center for Compulsive Behavior and Addiction, Rush University Medical Center, Chicago, IL, USA
⋅3 Department of Neurological Sciences, Rush University Medical Center, Chicago, IL, USA
The blood brain barrier (BBB) is a highly dynamic interface between the central nervous system (CNS) and periphery. The BBB is comprised of a number of components and is part of the larger neuro(glio)vascular unit. Current literature suggests that psychostimulant drugs of abuse alter the function of the BBB which likely contributes to the neurotoxicities associated with these drugs. In both preclinical and clinical studies, psychostimulants including methamphetamine, MDMA, cocaine, and nicotine, produce BBB dysfunction through alterations in tight junction protein expression and conformation, increased glial activation, increased enzyme activation related to BBB cytoskeleton remodeling, and induction of neuroinflammatory pathways. These detrimental changes lead to increased permeability of the BBB and subsequent vulnerability of the brain to peripheral toxins. In fact, abuse of these psychostimulants, notably methamphetamine and cocaine, has been shown to increase the invasion of peripheral bacteria and viruses into the brain. Much work in this field has focused on the co-morbidity of psychostimulant abuse and human immunodeficiency virus (HIV) infection. As psychostimulants alter BBB permeability, it is likely that this BBB dysfunction results in increased penetration of the HIV virus into the brain thus increasing the risk of and severity of neuro AIDS. This review will provide an overview of the specific changes in components within the BBB associated with psychostimulant abuse as well as the implications of these changes in exacerbating the neuropathology associated with psychostimulant drugs and HIV co-morbidity.
Introduction
The blood brain barrier (BBB) is a specialized structure formed by brain endothelial cells (BECs) that are tightly interconnected to form a boundary between the central nervous system (CNS) and periphery (Abbott et al., 2010). Once thought to be static, the BBB is now known to be an active and dynamic interface that responds to signals from both the brain parenchyma and vasculature. BBB dysfunction contributes to the pathophysiology of numerous CNS disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis, ischemia, and neuroAIDS (Stewart et al., 1992; Nath et al., 2001; Samii et al., 2004; Compston and Coles, 2008; Yenari and Han, 2012). Recent evidence points to the ability of drugs of abuse (e.g., stimulants) to disrupt BBB function. This review will provide an overview of normal BBB structure and function, focusing on the regulation of neuroinflammation, and will provide examples of BBB disruption specifically associated with stimulant drug abuse. As neuroAIDS is exacerbated by stimulant abuse, we will also discuss the implications of stimulant-induced BBB dysfunction as it relates to neuroAIDS disease progression.
Components of the BBB
The structure of the BBB has been thoroughly described in other reviews (Sandoval and Witt, 2008; Carvey et al., 2009). In brief, the BBB is comprised of highly specialized BECs which interact with pericytes, the vascular basement membrane, and astrocytes (Diaz-Flores et al., 1991; Krueger and Bechmann, 2010). BECs are closely associated through cell-cell complexes of tight junction proteins: claudins, occludins, and junctional adhesion molecules (JAMs; Hawkins and Davis, 2005). Adherens junctions also form between BECs. Accessory proteins like zona occludins (ZO), cingulin, and afadin (AF-6) provide structural support and stability to both tight and adherens junctions (Yamamoto et al., 1999; Mark and Davis, 2002). Though little is known about pericyte involvement within the BBB, these cells likely regulate cerebral blood flow (Bandopadhyay et al., 2001) and stabilize forming vessels (Ramsauer et al., 2002). BECs and pericytes are enveloped by extracellular matrix proteins that comprise the basement membrane (Farkas and Luiten, 2001). Astrocytes interact with components of the BBB through their foot processes that make contact with BECs to form the glia limitans perivascularis as well as the basement membrane (Krueger and Bechmann, 2010). Astrocytic foot process in the margin of the brain form a thin membrane called the glia limitans superficialis in the subarachnoid space. These two glia limitans are classically viewed as continuous, starting at the subarachnoid space, and merging with the parenchymal limitans as vessels penetrate into brain. Astrocytes facilitate BBB development by inducing tight junction formation, providing microvascular support, and allowing molecule (e.g., water, sugar, ions) diffusion into the brain (Janzer and Raff, 1987; Madsen and Hirschberg, 2010).
Neurovascular Coupling at the BBB Interface
There is a complex relationship between vascular and neuronal systems within the CNS. These interactions are promoted by the BBB, neurons, and glia, which together make up the neuro(glio)vascular unit (Hawkins and Davis, 2005; Koehler et al., 2009). This neurovascular coupling regulates cerebral blood flow enabling efficient oxygen and nutrient supply to various cell types and brain regions (Iadecola, 2004). Moreover, intracellular communication within the neuro(glio)vascular unit allows for localized control of cerebral blood flow to match the needs of specific brain regions. These interactions are important for blood flow to the neurons, as well as for the development, and maintenance of the neurovasculature.
Functions of the BBB
The main function of the BBB is to maintain homeostasis of the brain. This barrier protects the CNS from peripheral toxins within the circulation. These toxins may be endogenous factors or exogenous xenobiotics (Abbott et al., 2010). The impenetrability of the BBB results from physical restriction due to tight junction connectivity of BECs, transport regulation limiting transcellular migration, and enzymatic activities of BECs that metabolize harmful substances at the vascular face of the BBB (Madsen and Hirschberg, 2010).
Transport of molecules across the BBB is strictly regulated. A wide range of lipid-soluble molecules passively diffuse through BECs and certain molecules and cells are capable of paracellular migration between adjacent BECs. Most molecules are restricted either by lipid-solubility, polarity, or size, and thus require transporters to cross the BBB. Transporters found within the BBB are numerous, but in general, transporters can be categorized as being ion transporters, active transporters (e.g., P-glycoprotein), sodium-dependent transporters, or sodium-independent transporters (Carvey et al., 2009). These transport systems supply nutrients to the brain from the blood and remove metabolic byproducts from the brain to the blood for elimination (Ohtsuki, 2004).
Through neurovascular coupling, the BBB regulates cerebral blood flow. Contractile proteins identified in pericytes are thought to alter vascular diameter and cerebrovascular blood flow (Bandopadhyay et al., 2001). Transport systems bring in vasoactive factors from the periphery that interact with smooth muscle cells and pericytes surrounding the BBB endothelium to allow for vasodilation or constriction (Peppiatt et al., 2006; Bell et al., 2010). Additionally, cells within the neuro(glio)vascular unit can secrete local vasoregulators like norepinephrine, nitric oxide, and endothelin which regulate regional cerebral blood flow (Lecrux and Hamel, 2011).
Inflammatory cells and signaling factors from the periphery into the CNS are trafficked across the BBB. The neuroinflammatory responses produced by these cells and factors can also alter BBB function and stability. While it is yet to be determined whether BBB dysfunction occurs prior to neuroinflammation or vice versa, studies have clearly shown that the inflammatory response by resident neuroglia perpetuates BBB dysfunction and this BBB dysfunction increases the neuroinflammatory response. Under normal physiological conditions, the BBB limits the entry of most leukocytes into the CNS. However, select leukocytes like neutrophils, can cross the BBB during normal immune surveillance with little disruption to BBB tight junctions (Petty and Lo, 2002). During a neuroinflammatory response, immune factors, and endothelial adhesion molecules signal to increase immune cell migration through the BBB (Petty and Lo, 2002). Signals resulting in increased immune cell migration can alter the structural organization of tight junction proteins and lead to actin cytoskeleton remodeling in the BBB basement membrane (Deli et al., 1995; Couraud, 1998; Ransohoff et al., 2003). Migrating peripheral immune cells disrupt normal BBB function, and produce cytokines and chemokines within the CNS which furthers BBB disruption (Cartier et al., 2005). Cytokines [e.g., tumor necrosis factor alpha (TNFα), interleukin (IL)-1β and IL-6] and the CCL2 chemokine are increased in serum, neuronal tissue, and cerebrospinal fluid in several CNS disorders including traumatic brain injury (Morganti-Kossman et al., 1997), HIV-associated encephalitis (Cartier et al., 2005), and Huntington disease (Stolp and Dziegielewska, 2009). These neuroinflammatory factors likely contribute to BBB breakdown occurring in these disorders through activity at their receptors on BECs and other BBB cell types (Buckner et al., 2006). Peripheral immune cells can also trigger increased production of inflammatory factors (Verma and Szmitko, 2006; Fletcher et al., 2009), neurotransmitters (e.g., glutamate), neurotrophic factors (e.g., vascular endothelial growth factor), and proteases (e.g., matrix metallopeptidase (MMP-9; Petty and Lo, 2002) from BECs and astrocytes. These factors perpetuate neuroinflammation, and contribute to continuing alterations to the BBB (Petty and Lo, 2002). Alterations in BBB integrity resulting from neuroinflammation produce a cascade resulting in further BBB disruption and increased penetration of immune cells into the CNS. This feed-forward cycle can lead to disruptions in physiological functions of the BBB normally in place to protect against peripheral toxins and xenobiotics, allow for cross-barrier transport, and regulate cerebral blood flow.
Many abused drugs, including stimulants, are pro-inflammatory which leads to BBB disruption and propagates the cyclic relationship between neuroinflammation and BBB damage. This may accelerate the onset of the neurotoxicities associated with chronic use of these potent drugs (Czub et al., 2001; Nath et al., 2002). This review focuses on the effects of psychostimulant drugs though many of the molecular and cellular mechanisms of BBB dysfunction and neuroinflammation have been well-characterized in studies focused on the CNS depressant alcohol (see reviews: de la Monte et al., 2009; Perdisky et al., 2011; Strazza et al., 2011).
Stimulant Drugs and the BBB
Methamphetamine
Current surveys estimate that approximately 13 million people ages 12 and older have abused methamphetamine (meth) in their lifetimes, making it the second most widely abused illicit drug in the United States after cannabis (NSDUH 2011; United Nations Office on Drugs and Crime 2007). Meth reverses the transport of norepinephrine, dopamine (DA), and serotonin (5-HT) leading to excess release of these monoamines from the cytoplasm and storage vesicles into the synapse (Rothman et al., 2001). Meth also prevents monoamine reuptake causing them to remain in the synaptic cleft to increase post-synaptic receptor stimulation. Chronic meth abuse in humans is associated with neurotoxicities resulting in damage to both DA and 5-HT terminals in a variety of brain regions (see review: Krasnova and Cadet, 2009). Proposed mechanisms underlying meth-induced neurotoxicity include increased reactive oxygen species (ROS) and dopamine-quinone (DAQ) production (Kuhn et al., 2006), hyperthermia (Kiyatkin et al., 2007), neuroinflammation (Guilarte et al., 2003), and BBB dysfunction (Sharma and Kiyatkin, 2009). It is likely that more than one mechanism underlies the neuronal maladaptations and damage associated with meth abuse, and that this damage is associated with the interaction of multiple mechanisms. For example, oxidative stress and hyperthermia can increase BBB dysfunction which in turn, can exacerbate neuroinflammation after meth exposure. While some studies show direct neurotoxicity (Ricaurte et al., 1980; McCann et al., 2008) after meth exposure without BBB disruption or neuroinflammation, these studies did not assess changes in the BBB or in the inflammatory response so it is uncertain whether neurotoxicity can occur independent of BBB disruption and/or neuroinflammation.
Preclinical studies have reported BBB dysfunction in both whole animal and in vitro models. Our laboratory recently showed that a single intraperitoneal injection of either 3 or 9 mg/kg meth in rats resulted in BBB damage in the prefrontal cortex and nucleus accumbens shell, identified by punctate areas of fluorescein isothiocyanate (FITC)-labeled albumin leakage (Kousik et al., 2011). Other rodent studies also showed BBB dysfunction following acute meth treatment marked by leakage of Evans blue into the parenchyma or through increased IgG immunostaining (Bowyer and Ali, 2006; Bowyer et al., 2008;Kiyatkin and Sharma, 2009, 2011; Sharma and Kiyatkin, 2009; Kuroda et al., 2010; Martins et al., 2011). The treatment regimens varied greatly from single doses ranging from 3-40 mg/kg to several acute doses over 24 h, all of which compromised BBB integrity. Alterations to the BBB occurred where meth-induced changes in DA or 5-HT levels and terminal damage were most pronounced, including the striatum, cortex, and hippocampus, but not in areas where monoamine levels were unaffected by meth (e.g., the substantia nigra; Sharma and Ali, 2006; Bowyer et al., 2008; Kousik et al., 2011; Martins et al., 2011). Increased permeability across BECs after meth treatment was also observed in vitro through dose-dependent decreases in transendothelial electrical resistance, a method commonly used to measure paracellular permeability (Mahajan et al., 2008; Zhang et al., 2009; Abdul Muneer et al., 2011). Clinical studies report increased levels of peripheral toxins, like HIV and hepatitis C virus, in the brain parenchyma of meth-abusing humans (Nath et al., 2001;Letendre et al., 2005, 2007; Schep et al., 2010). In fact, 70% of individuals undergoing treatment for hepatitis C in one study were self-reported chronic meth abusers (Letendre et al., 2007). These individuals had a higher hepatitis C viral load in the brain than meth-naïve individuals diagnosed with hepatitis C suggesting that meth-induced BBB dysfunction can increase viral penetration into the CNS (Letendre et al., 2007).
Alterations in tight junction complexes can contribute to loss of BBB integrity after meth treatment. Meth treatment decreases the expression of ZO-1, occludin, and claudin-5 in both in vivo and in vitro studies (Mahajan et al., 2008; Ramirez et al., 2009; Banerjee et al., 2010; Abdul Muneer et al., 2011; Martins et al., 2011). Additionally, meth can also alter other BBB cell types and interactions. For example, meth increases the expression of peptidases, like MMP-1 and MMP-9, which are involved in degrading certain tight junction proteins to produce structural changes to the BBB basement membrane (Conant et al., 2004). Meth also activates microglia and astrocytes within the neuro(glio)vascular unit which may contribute to meth-induced neurotoxicity, potentially through the secretion of inflammatory cytokines, chemokines, and vasoactive factors (Kiyatkin et al., 2007; Wisor et al., 2011). These structural and functional changes within the BBB/neuro(glio)vascular unit can also result in brain edema and disruption of proper ion flow across the barrier (Sharma and Kiyatkin, 2009).
Neuroinflammation is a proposed mechanism underlying meth-induced neurotoxicity and may also contribute to BBB damage produced by meth. A single 30 mg/kg intraperitoneal injection of meth increases the expression TNFα and IL-6 in the hippocampus, frontal cortex, and striatum in mice (Goncalves et al., 2008). Similar increases in TNFα, IL-1β, and IL-6 are reported after multiple meth treatments and are potentially linked to meth-induced microglial activation (Cadet et al., 1994; Lai et al., 2009; Goncalves et al., 2010). Mahajan et al. (2008) showed that exposure to 50 nM meth produced a 47% increase in the transmigration of peripheral blood mononuclear cells using an in vitro BBB model. Meth-induced increases in glial activation as well as TNFα and TNF receptor expression were attenuated with pre-treatment of indomethacin, an anti-inflammatory agent, suggesting a potential therapeutic target for impeding meth-neurotoxicity (Goncalves et al., 2010). In summary, neuroinflammation contributes to the neurotoxicities seen after meth exposure and potentiates the feed-forward cycle between neuroinflammation and BBB damage to produce prolonged BBB damage (see Table 1 for a summary of the effects of meth on BBB function).
MDMA (ecstasy)
3,4-Methylenedioxymethamphetamine (MDMA or “Ecstasy”) is a synthetic derivative of amphetamine. There is a growing population of MDMA abusers, particularly among young adults who attend “raves” or private clubs (NIDA 2008). Like other amphetamines, MDMA increases the release of monoamines from nerve terminals through interactions with the serotonin transporter (SERT) and dopamine transporter (DAT; Sulzer et al., 2005). MDMA has a higher affinity for SERT than DAT resulting in a greater release of 5-HT than DA (Rothman and Baumann, 2003). Like meth, secondary release of glutamate may contribute to the neurotoxic effects of MDMA (Nash and Yamamoto, 1992). Though acute consequences of MDMA use have been well-studied, long-term effects and potential neurotoxicities are not well known. MDMA appears to be selectively toxic to 5-HT terminals and produces long-lasting depletions of 5-HT in the hippocampus, prefrontal cortex, amygdala, and striatum (Morgan and Gibb, 1980; Ricaurte et al., 1985, 1988). Moreover, thioester metabolites of MDMA, including 3,4-dihydroxyamphetamine (DHA), also produce long-term depletion of 5-HT (Monks et al., 2004). Though 5-HT toxicities have been identified in multiple brain areas, the mechanisms underlying this selective toxicity remain unknown.
Recent studies suggest that BBB dysfunction may contribute to MDMA-induced neurotoxicities. Increased leakage of Evans Blue or trypan blue as well as increased IgG immunostaining were reported in several cortical areas, hippocampus, cerebellum, and striatum of rats treated acutely with MDMA (Yamamoto and Bankson, 2005; Sharma and Ali, 2008; Torres et al., 2011). BBB dysfunction was observed immediately following acute MDMA treatment and up to 10 weeks following an acute injection. Increased BBB permeability after MDMA treatment was associated with increased parenchymal penetration of endogenous albumin (Sharma and Ali, 2008), increased activation of astrocytes, and microglia (Monks et al., 2004), and increased brain water content suggesting edema (Sharma and Ali, 2008).
In addition to altering BBB permeability, MDMA increases expression of pro-inflammatory cytokines, including IL-1β, in brain tissue (Torres et al., 2011). These cytokines can contribute to the feed-forward cycle between BBB dysfunction and neuroinflammation. MDMA abuse also leads to oxidative stress (Yamamoto and Bankson, 2005). MDMA-induced excess release of DA and 5-HT results in the formation of ROS, DAQ, and toxic metabolites from 5-HT oxidation (Quinton and Yamamoto, 2006). The accumulation of toxic free radicals increases the susceptibility of brain tissue to ischemic injury and triggers a variety of signaling cascades leading to BBB dysfunction, brain edema, and neuroinflammation (Gu et al., 2012). It is likely that the oxidative stress and neuroinflammation produced by MDMA results in BBB dysfunction, though further research is needed to verify if MDMA has a direct detrimental effect on the BBB (see Table 2 for a summary of the effects of MDMA on BBB function).
Cocaine
Cocaine inhibits monoamine reuptake, particularly through binding DAT (Barnett et al., 1981). While cocaine abuse was at its highest levels in the 1970-1980s, the United States is still the world’s number one importer and user with approximately 1.9 million current cocaine abusers (NSDUH 2008). Cocaine abuse is not linked to DA or 5-HT terminal damage (Bennett et al., 1993) though cocaine can produce oxidative stress and neuroinflammation. These potentially toxic effects likely contribute to cocaine-induced BBB dysfunction. Chronic cocaine administration (30 mg/kg/day intraperitoneal injection) ruptures the neurovascular capillaries and basement membranes of rats (Barroso-Moguel et al., 1997). Increased BBB permeability marked by approximately 50% more Evans Blue or sodium fluorescein leakage from blood to brain after cocaine exposure has also been observed in rodents (Sharma et al., 2009; Yang et al., 2010; Yao et al., 2011). Whole animal studies are more recent in this field as most studies investigating the effects of cocaine on BBB integrity use an in vitro BBB model comprised of brain microvascular endothelial cells and C6 astrocytes. Decreases in transendothelial electrical resistance and increases in FITC-Dextran leakage across an endothelial monolayer indicates increased permeability across BECs after cocaine treatment (Fiala et al., 1998, 2005; Zhang et al., 1998; Gandhi et al., 2010; Yao et al., 2011). Clinical studies reveal similar BBB breakdown in the basal ganglia and increased HIV penetration into the CNS of cocaine-abusing humans (Nath et al., 2001).
Cocaine-induced BBB dysfunction is partially characterized by loss of, or alterations in, tight junction protein complexes (Fiala et al., 2005, 2008). Significant decreases in ZO-1 and JAM-2 occur in both rodents and in vitro BBB preparations (Dhillon et al., 2008; Gandhi et al., 2010; Yao et al., 2011). Cocaine treatment also increases gene expression of factors, including MMP-1, that contribute to basement membrane actin rearrangement resulting in stress fiber formation around cerebral vessels (Nair et al., 2004; Fiala et al., 2005; Dhillon et al., 2008). Persistent loss of, or conformation changes in tight junction proteins, and reorganization of basement membrane fibers leave the brain open to peripheral toxin penetration leading to CNS disorders linked with cocaine abuse (see Table 3 for a summary of the effects of cocaine on BBB function).
Neuroinflammation induced by cocaine (through increases in endothelial adhesion molecules and inflammatory factors) potentially plays an important role in BBB dysfunction. Increases in intracellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), endothelial-leukocyte adhesion molecule (ELAM or selectin-1), and platelet endothelial cell adhesion molecule 1 (PECAM1), have been indentified in both preclinical studies (Fiala et al., 1998, 2005; Gan et al., 1999; Chang et al., 2000) and in cocaine-abusing humans (Buch et al., 2011). Increases in circulating leukocytes and cell adhesion molecules on leukocytes have also been reported with cocaine treatment in rodents and in vitro (Chang et al., 2000; Nair et al., 2004). Increases in cell adhesion molecules on the BBB endothelium and on leukocytes promote more leukocyte extravasation from blood to brain (Barroso-Moguel et al., 1997; Gan et al., 1999). Indeed, cocaine treatment in vitro results in a 100% increase in the number of peripheral blood mononuclear cells migrating across BECs (Fiala et al., 1998, 2008; Zhang et al., 1998). Cocaine-induced increases in leukocyte-endothelial adhesion are accompanied by elevated levels of pro-inflammatory cytokines and chemokines. Western blotting and immunostaining reveal increases in the expression of TNFα, IL-6, IL-8, nuclear factor kappa B (NFκB), activator protein 1 (AP-1), and CCR2 (Zhang et al., 1998; Gan et al., 1999; Lee et al., 2001; Dhillon et al., 2008). These inflammatory signals are also associated with increased viral invasion of macrophage-tropic HIV across an in vitro BBB (Fiala et al., 1996; Zhang et al., 1998) which may explain the greater neuropathology observed in patients co-morbid for HIV infection and cocaine abuse (Fiala et al., 2008). The effects of cocaine on the BBB are likely exacerbated by cocaine-induced neuroinflammation.
Nicotine
Though the number of cigarette smokers in the United States has been steadily on the decline since the 1960s, an estimated 45.3 million Americans are still considered daily cigarette smokers (CDC 2010). An average cigarette yields approximately 1 mg of absorbed nicotine, a stimulant alkaloid responsible for the addictive-properties of cigarette smoking (Connolly et al., 2007). Nicotine is a soluble small molecule that rapidly diffuses through the BBB and can interact directly with nicotinic acetylcholine receptors (nAChRs) on BECs (Le, 2003). Preclinical studies using rodent models of acute and chronic nicotine exposure reported compromised BBB integrity, marked by Evans blue or [14C] sucrose leakage into brain parenchyma. This BBB disruption occurred as early as 2 h after acute nicotine and after 6 weeks of chronic exposure (Lin et al., 1992; Uzum et al., 1999; Venisnik, 2000;Hawkins et al., 2004, 2005). In vitro studies using either brain microvascular endothelial cells alone or in combination with C6 astrocytes revealed increases of FITC-Dextran and [14C]sucrose leakage across the cells after nicotine treatment (Schilling et al., 1992; Abbruscato et al., 2002). Nicotine also decreases transendothelial electrical resistance in an in vitro BBB model (Hutamekalin et al., 2008; Rodriguez-Gaztelumendi et al., 2011). This decrease in resistance is dose-dependent (0.1-100 μM) and sufficient enough to produce a 60-150% increased invasion of Escherichia coli across brain microvascular endothelial cells (Chen et al., 2002). Whole animal studies also revealed nicotine-induced BBB dysfunction leading to increased transport from blood to brain of xenobiotics like squanivir, a protease inhibitor included in anti-retroviral therapy for HIV infection (Manda et al., 2010a). These studies offer added insight into the consequences of nicotine use relating to uptake of therapeutics into the CNS. Finally, both in vitro and in vivo studies report significant loss, or alterations in, tight junction proteins including ZO-1, claudin-3, JAMs, and occludin (Abbruscato et al., 2002; Hawkins et al., 2004; Hutamekalin et al., 2008; Manda et al., 2010b).
In addition to altering BBB tight junctions, nicotine affects transport and receptor systems involved in normal BBB function. Nicotine increases the activity of monocarboxylase and organic cation transporters while decreasing the functional activity of ion transporters like Na+, K+, 2Cl− co- transporter, and Na+, K+-ATPase on BECs (Wang et al., 1994; Abbruscato et al., 2004; Lockman et al., 2005; Paulson et al., 2006; Liou et al., 2007). Nicotine treatment produced a 22 and 17% decrease in Na+ and K+-ATPase activity in the microvasculature and brain, respectively (Wang et al., 1994). Nicotine also inhibited the activity of P-glycoprotein, an important efflux transporter responsible for impeding the entry of a range of compounds into the CNS (Manda et al., 2010a). Changes in transport mechanisms across the BBB can produce detrimental alterations in ion gradients and in the nutrients available to the brain (Paulson et al., 2006; Yang et al., 2006). Nicotine-induced alterations in ion transport are especially injurious as changes in ion gradients lead to brain edema lasting for up to 3 weeks after nicotine exposure as well as increased cerebral ischemic injury (Wang et al., 1997; Paulson et al., 2010). nAChRs on cells within the BBB are also affected by nicotine. Chronic nicotine decreases nAChR surface expression as well as expression of certain nAChR subunits, particularly the α2 isoform, on BECs (Wang et al., 1994; Abbruscato et al., 2004; Lockman et al., 2005). While nicotine activity at nAChRs on BECs alters the BBB, treatment with nAChRs antagonists (e.g., mecamylamine, α-bungarotoxin, and hexamethonium) decreases nicotine-induced BBB dysfunction. This decrease in BBB disruption was identified by attenuation of [14C]sucrose leakage into the brain parenchyma, reversal of ZO-1 loss, and decreased Escherichia coli invasion in an in vitro BBB model (Abbruscato et al., 2002; Chen et al., 2002; Conklin et al., 2002; Hawkins et al., 2005; see Table 4 for a summary of the effects of nicotine on BBB function).
Nicotine can activate inflammatory signals that directly affect BBB integrity. Studies have highlighted the effects of nicotine on adhesion molecules necessary for immune cell extravasation from the blood into the brain. Nicotine enhances peripheral blood mononuclear cell migration across cerebral vessels through a cascade promoting increased expression of adhesion molecules and inflammatory cytokines (Yong et al., 1997; Albaugh et al., 2004; Bradford et al., 2011). The expression of ICAM-1, VCAM-1, and P-selectin is increased with chronic nicotine treatment (Yong et al., 1997; Albaugh et al., 2004). Furthermore, nicotine increases gene expression of TNFα, IL-18, IL-1β, and chemokines including CCL2, CCL8, and CXC3CL1 (Bradford et al., 2011). Nicotine also decreases gene expression of anti-inflammatory factors (e.g., Bcl6, IL-10, and CCL25; Bradford et al., 2011). These studies indicate that nicotine increases the expression of inflammatory cytokines and endothelial adhesion molecules and can enhance leukocyte migration across the BBB, particularly during neuroinflammation.
Stimulant Co-Morbidity with HIV Infection
Mechanisms thought to underlie stimulant-mediated toxicities parallel those associated with HIV infection. Indeed, the co-morbidity of stimulant abuse and HIV is well documented. Studies report that meth abuse is linked with enhanced frequency of unprotected sex and increased HIV transmission (Semple et al., 2004; Buchacz et al., 2005). Moreover, the morbidity and mortality of meth-abusing HIV-positive individuals far exceeds that seen with either pathology alone.
HIV infects peripheral leukocytes which migrate across the BBB, thus allowing the virus access to the CNS (An et al., 1999). This migration occurs early in HIV infection (Risdahl et al., 1998; Romanelli et al., 2006) though HIV-induced toxicity of BECs occurs throughout infection and parallels HIV toxicity of neurons (Price et al., 2005). Once HIV-infected cells enter the brain, HIV-produced proteins trigger neuroinflammation which can exacerbate the neuroinflammatory effects of stimulant drugs. As overviewed above, stimulant drug abuse compromises BBB integrity through decreases in tight junction proteins and increases in endothelial adhesion molecules which promote leukocyte extravasation from the blood into the brain.
Meth increases the expression of HIV co-receptors, CXCR4 and CCR5 (Nair et al., 2009), and can significantly increase HIV replication in astrocytes (Gavrilin et al., 2002). In HIV-positive meth abusers, meth-induced BBB dysfunction facilitates increased transport of HIV-infected leukocytes into the brain (Liang et al., 2008). Moreover, HIV-induced BBB damage might increase the concentration of meth within the CNS contributing to further meth-neurotoxicity. Co-treatment of meth and HIV protein gp120 decreases transendothelial electrical resistance across BECs more than treatment with meth or gp120 alone (Mahajan et al., 2008). Administration of meth and gp120 or the HIV protein Tat in combination, further decreased the expression of ZO-1, claudin-3, claudin-5, and JAM-2 than either treatment alone (Mahajan et al., 2008; Banerjee et al., 2010). In addition to the effects of meth and HIV on tight junction proteins, increased levels of MMP-2 and MMP-9 were reported with meth and Tat treatment in an in vitro BBB model (Conant et al., 2004) and in the cerebrospinal fluid of HIV-positive individuals (Conant et al., 1999; Liuzzi et al., 2004). These peptidases can produce persistent BBB damage, leaving the brain open to prolonged infiltration of HIV and other peripheral toxins.
Clinical investigations revealed potential additive and/or synergistic effects of meth and HIV within the CNS. Chang et al. (2005) found increased neuronal loss in frontal gray matter (−5.7%), frontal white matter (−6.1%), and basal ganglia (−6%) when compared with HIV-positive meth-naïve individuals and HIV-negative meth abusers. These studies also reported increased glial activation and changes in cellular metabolism in frontal white matter and basal ganglia in HIV-positive meth abusers (Chang et al., 2005). Meth-induced glial activation may lead to increased progression of neuroAIDS in HIV-positive meth abusers (Garden, 2002). DA neurotoxicity is associated with both meth abuse, and HIV infection appears worsened by the combination (Czub et al., 2001; Maragos et al., 2002; Theodore et al., 2006). Increased DA neurotoxicity with meth abuse and HIV/AIDS co-morbidity may increase the risk of DA-associated disorders like PD. In fact, meth abusers have a twofold increase in developing PD compared with healthy controls or cocaine abusers (Callaghan et al., 2012) and as many as 10% of patients with HAND also display Parkinsonian features (Berger and Nath, 1997). In summary, the neuropathological features associated with both meth abuse and HIV/AIDS may be exacerbated through meth-induced BBB dysfunction resulting in, or worsened by, neuroinflammation.
Similar to meth abuse, individuals abusing MDMA are more likely to engage in risky sexual behaviors and injection drug use (Dunn et al., 2010). Indeed, 77% of participants in a clinical investigation reported not using condoms when under the influence of MDMA, and of this group, 54% reported having two or more sexual partners during this time (Dunn et al., 2010). In a similar clinical study, 9.3% of MDMA users surveyed reported having unprotected sex with partners who were either HIV-positive or of unknown HIV status (Klitzman et al., 2002). These behaviors likely increase HIV transmission within this population (Mitcheson et al., 2008). Little is currently known about the neurotoxic implications of MDMA and HIV co-morbidity. As MDMA produces BBB dysfunction as well as neuroinflammation and oxidative stress, which both perpetuate BBB damage, MDMA-induced BBB dysfunction would be expected to increase HIV-infected leukocyte infiltration into the CNS. The interaction between MDMA and HIV has not yet been characterized and may represent an unknown population at risk for HIV and MDMA-associated neurotoxicities. These toxicities may be exacerbated by MDMA-induced BBB dysfunction leading to increased HIV penetration into the brain.
Early investigations reported cocaine abuse as a risk factor for increased HIV transmission and a more rapid progression to AIDS (Fiala et al., 1998; Webber et al., 1999). Cocaine increases HIV infiltration into the CNS through increased HIV-infected leukocyte adhesion and transmigration across the BBB endothelium. BBB dysfunction observed following cocaine exposure can increase the capacity of other immune cells and inflammatory factors to enter into the brain from the periphery. These features of cocaine likely promote the neuroinflammation associated neuroAIDS. Cocaine also increases HIV replication in monocytes, macrophages, and astrocytes (Peterson et al., 1991; Roth et al., 2002; Reynolds et al., 2006). This is especially important as astrocytes may act as a reservoir for HIV within the brain (Nath, 2010). Preclinical studies revealed that cocaine enhances oxidative stress, neuronal dysfunction, and apoptosis in gp120 and Tat-treated cells in vitro and in rodents (Koutsilieri et al., 1997; Nath et al., 2000; Bagetta et al., 2004). Recent findings from Napier et al. (2010) revealed that cocaine and Tat synergize to over activate cortical neurons. Like cocaine, Tat can inhibit DAT which may contribute to HIV-induced DA neurotoxicity (Aksenov et al., 2008). When combined, cocaine plus Tat may result in DA neurotoxicities which exacerbate HIV neuropathology. Cocaine-induced BBB dysfunction leading to increased HIV transmission into the CNS is apt to contribute to the resulting neurotoxicities associated with cocaine and HIV co-morbidity.
There are a number of clinical reports documenting the effects of cocaine and HIV co-morbidity. Cocaine abuse accelerates HIV disease progression by decreasing CD4+ T-cell counts and increasing HIV viral load, independent of anti-retroviral therapies (Baum et al., 2009). Chaisson et al. (1989) found that daily cocaine use increased the risk of HIV infection up to sixfold in African American and Hispanic populations. Persistent cocaine abusers are also three times more likely to die from AIDS-related complications than cocaine-naïve individuals (Cook et al., 2008). This suggests a synergistic interaction between cocaine and HIV which exacerbates HIV disease progression. Interactions between cocaine and HIV within the brain parenchyma may be enhanced through cocaine-induced increases in HIV-infected leukocyte trafficking across the BBB.
Studies report a 51% or higher cigarette smoking rate among nationally surveyed HIV-positive individuals (Collins et al., 2001; Gritz et al., 2004; Burkhalter et al., 2005). Though little is known about the effects of nicotine on HIV disease progression, smoking increases the likelihood of HIV-related complications (e.g., bacterial pneumonia and HIV-associated dementia) and increases the mortality rate of HIV-positive individuals (Hirschtick et al., 1995; Conley et al., 1996; Feldman et al., 2006). Furthermore, nicotine can act as a major immunosuppressive agent by attenuating the immune and virological response to anti-retroviral therapies by up to 40% (Humfleet et al., 2009). Nicotine has both pro- and anti-inflammatory properties. The immunosuppressive effects of nicotine include increasing T-cell unresponsiveness, increasing IL-4 production, and inhibiting the production of certain pro-inflammatory cytokines (Sopori and Kozak, 1998). Immunosuppressive functions can add to the already detrimental immune suppression produced by HIV infection leading to an accelerated progression of HIV-associated complications. The pro-inflammatory effects of nicotine within the CNS include increasing the release of pro-inflammatory cytokines and increasing leukocyte transmigration through BECs from blood to brain. Nicotine may increase HIV viral load within the CNS through increasing BBB permeability and promoting leukocyte migration into the brain parenchyma. Nicotine enhances the production of HIV in macrophages (Abbud et al., 1995) and in activated microglia (Rock et al., 2008). Similar increases in HIV replication are also seen with opiate and cocaine abuse which are reported to promote HIV neuropathology (Nath et al., 2002; Roth et al., 2005). The scope of what is currently known about the effects of nicotine on HIV disease progression is limited, though HIV-positive individuals who smoke cigarettes show a decreased immune response, poorer responses to anti-retroviral therapies, greater risk of viral rebound, and an increased probability for HIV-related complications (Feldman et al., 2006). Nicotine may increase HIV infiltration into the CNS through nicotine-induced BBB dysfunction and increase HIV replication within microglia in the brain to accelerate progression to neuroAIDS.
Conclusion
The BBB/neuro(glio)vascular unit plays an important function in the neurotoxicity of stimulant abuse; both as a factor involved in the development of neurotoxicities and also in its response to these toxic effects. This suggests that components within the BBB and neuro(glio)vascular unit may be potential therapeutic sites targeted to impede the progression of stimulant-induced neurotoxicity. Most stimulants dysregulate the BBB through alterations in tight junction complexes or through oxidative stress and neuroinflammation. Neuroinflammation plays a particularly important role as it contributes to a feed-forward process between BBB dysfunction and neuroinflammation which likely results in the CNS being more vulnerable to infiltration of pathological mediators from the periphery. This is especially important in co-morbid situations of stimulant abuse and immune disorders, like HIV/AIDS. Stimulants not only increase the migration of peripheral HIV-infected leukocytes into the CNS, but once inside the brain, can accelerate the progression to neuroAIDS.
The effects of stimulant drugs have been extensively researched, though further studies are required to advance the field toward identification of therapeutic targets for medication development. Preclinical studies thus far have revealed a great deal about the direct and indirect effects of stimulants on the BBB. Future studies must build upon what is already known using a model of stimulant abuse and co-occurring morbidities, like HIV infection. Such models of co-morbidity would better emulate the human condition and would allow more direct assessments of the dynamic interactions between all cells within the BBB/neuro(glio)vascular unit in these clinically important human scenarios.
|
|
| |
| |
|
|
|