 |
|
|
| |
The 18th International Workshop on Clinical Pharmacology of Antiviral Therapy - Report by Courtney Fletcher, Pharm. D., University of Nebraska Medical Center
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
Jennifer J. Kiser, Pharm.D.
Associate Professor
School of Pharmacy
University of Colorado at Denver
The 18th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy was held in Chicago, IL from June 14-16, 2017. In this report, we will highlight abstracts focused on pharmacologic issues that are of broad interest or might benefit from some expert clarification. Abstracts will be discussed in two general categories: (i) the clinical pharmacology of therapy for HIV infection and (ii) the clinical pharmacology of therapy for hepatitis C infection. This report does not cover all plenaries and data presented at the Workshop. For more information and to view presentations from the meeting please visit:
http://www.infectiousdiseasesonline.com/event/meeting-reports/antiviralpk-2017/
The Clinical Pharmacology of Therapy for HIV Infection.
Courtney V. Fletcher, Pharm.D.
A. Drug Development
1. In SWORD-1 and 2, DTG and RPV concentrations in persons switching from EFV or NVP are comparable with those switching from other ARVs (abstract O_10).
The SWORD studies presented at CROI 2017, (abstract 44LB) showed a switch from current ART in persons with HIV-RNA < 50cpm for 12 months to DTG+RPV once daily with food (N=513) was non-inferior to remaining on current ART (N=511): 95% of participants on both regimens maintained HIV-RNA < 50cpm. Of interest is whether persons switching from EFV and NVP, given these NNRTIs can reduce DTG and RPV concentrations, are at any risk of subtherapeutic DTG and RPV concentrations and early virologic failure. At 2 weeks after the switch, DTG and RPV concentrations were lower than those who did not switch from EFV or NVP, reflecting residual induction of hepatic metabolism; however, the DTG and RPV concentrations were above in vitro target concentrations. By week 4, DTG and RPV concentrations were comparable with those of the overall study population. These data provide reassurance to clinicians that persons receiving an EFV or NVP-containing regimen can be switched to the DTG+RPV combination without an increased risk of subtherapeutic concentrations.
Pharmacokinetics of Dolutegravir and Rilpivirine After Switching to the Two-Drug Regimen From an Efavirenz- or Nevirapine-Based Antiretroviral Regimen: SWORD-1 & -2 Pooled PK Analysis - (06/20/17)
2. A FDC tablet of DTG+RPV is bioequivalent with each drug given separately under fed conditions. (abstract P_25).
A regulatory filing of the DTG+RPV combination is planned. Abstract P_25 evaluated the bioequivalence in N=113 healthy volunteers of a fixed dose combination (FDC) tablet of DTG+RPV compared with each drug given separately. All drugs were given with a moderate fat meal. The FDC tablet was shown to be bioequivalent, and will increase the convenience of the DTG+RPV combination, assuming (a good bet) FDA approval.
Bioequivalence of a Fixed Dose Combination Tablet of Dolutegravir and Rilpivirine in Healthy Subjects - (06/20/17)
3. GSK2838232 (GSK-232), an HIV maturation inhibitor, is in early phase development in HIV-infected persons.
Maturation inhibitors work very late in the life-cycle of HIV to prevent the replication of HIV into a mature, infectious viral particle. GSK-232 is an investigational HIV maturation inhibitor in early clinical development. The drug is potent (see CROI 2015, abstract 538), highly protein bound, metabolized primarily by CYP3A4, and has enhanced bioavailability when given with food. RTV significantly boosts GSK-232 concentrations, with an approximate 10-fold increase in AUC and 3-fold increase in Cmax. To date, GSK-232 has been given to approximately 124 healthy persons with acceptable short-term tolerance. GSK-232 is moving forward in studies in HIV-infected persons, with a working dose of 50 mg once daily given with cobicistat; it is a novel drug that inhibits a step in the life-cycle of HIV that no currently approved drugs do.
GSK2578999, an HIV-1 maturation inhibitor with an improved virology profile against gag polymorphisms - (03/05/15)
Pharm Wk/2017: Early Safety, Tolerability and Pharmacokinetic Profile of GSK2838232, a Novel 2nd Generation HIV Maturation Inhibitor, as Assessed in Healthy Subjects - (06/20/17)
4. FDA perspectives on drug-drug interactions.
One of the strengths of this meeting is that it provides a venue for scientists from academia, industry and regulatory bodies to come together to discuss antiviral drug development. A hallmark of HIV drug development has been learning that drug-drug interactions can both preclude a drug from being developed and can be essential to a drug's development. Dr. Kellie Reynolds, deputy director of the FDA Division of Clinical Pharmacology IV, gave a superb plenary on the FDA perspectives on drug interactions and highlighted a new FDA guidance expected this year on concepts and conduct of drug-drug interaction studies. Her slides are available on the conference website given above. I am most hopeful to see an increased emphasis on management of drug-drug interactions (e.g., do not use; change dose; change frequency; stagger administration, etc.) and a move away from a recommendation of use with caution, which is not informative or very helpful to clinicians.
http://regist2.virology-education.com/2017/18AntiviralPK/16_Reynolds.pdf
B. HIV Pharmacotherapy.
1. A reappraisal of TAF vs. TDF nephrotoxicity?
Dr. Dario Cattaneo from Milan gave a very provocative oral abstract on the clinical trials of TAF (abstract O_2) and whether the renal safety benefit of TAF when compared with TDF was overestimated. The argument he presented goes as follows. First, TDF-related renal toxicity has been associated with higher tenofovir (TFV) plasma concentrations, arising, for example, from lower body weight or concomitant administration with ritonavir-boosted PIs. Second, Cattaneo and colleagues evaluated 510 HIV-infected persons receiving TDF-containing ART and separated them into four classes based on concomitant ARVs: PI/r, NNRTIs, INSTIs, and EVG/cobi. Plasma TFV concentrations were higher in those receiving TDF with PIs/r and EVG/cobi compared with NNRTIs and INSTIs, consistent with ritonavir or cobicistat inhibition of intestinal P-glycoprotein as the mechanism for increased plasma TFV concentrations. In these 510 persons, 149 discontinued TDF during a mean of 1149 days of follow-up. The probability of TDF discontinuation in the first year was significantly higher for those receiving EVG/cobi (43.6%) compared with 15.6% for PIs/r, 13.1% for NNRTIs and 10.6% for INSTIs. Cox hazard regression analysis found EVG/cobi and TFV plasma concentrations were both associated with an increased rate of TDF discontinuation. Third, the investigators noted that no dose adjustment of TDF is recommended when given with either ritonavir (e.g. PI/r) or cobicistat (e.g. EVG/cobi). However, when TAF is given with cobicistat the dose is reduced: TAF is given at 10mg once daily when given with cobicistat vs. 25 mg once daily if no PK booster (e.g. TAF combined with FTC and rilpivirine). The hypothesis advanced by these authors is in the clinical trials comparing TAF with TDF when both were combined with EVG/cobi/FTC, the TAF dose was reduced for concomitant administration with cobicistat (thereby adjusting for the drug-drug interaction) whereas there was no dose adjustment for TDF and that this lack of a dose adjustment led to higher rates of TFV-related nephrotoxicity in the TDF arm (as they observed in their retrospective analysis).
Some commentary. The pharmacologic basis for the argument is compelling, and it would be interesting to test this hypothesis in a prospective trial with a downward adjusted dose of TDF to compensate for the drug interaction. Such a study, however, is unlikely to ever be done. Thus, it is important to note that both TAF and TDF are quite well tolerated and rates of nephrotoxicity are low. In the two randomized studies of TAF vs. TDF, each with EVG/cobi/FTC, the increases in serum creatinine were 0.08 vs. 0.12 mg/dL, respectively, for TAF and TDF (Sax PE, Lancet 2015;385:2606).
A current and related conundrum that I believe does need clinical study is the safety of TAF at the 25 mg once daily dose (as found in the combination with FTC, Descovy) if given with a boosted PI, such as darunavir/cobicistat. If the TAF dose is downward adjusted from 25mg to 10mg once daily when given with EVG/cobi, should it really be 25mg once daily when given with DRV/cobi?
Effect of cobicistat on tenofovir disoproxil fumarate (TDF) durability: is it time to rethink at TAF trials? - (06/21/17)
2. Do not combine etravirine (ETR) with darunavir/cobicistat (DRV/c).
When darunavir/ritonavir (DRV/r) 600mg/100mg twice daily is given with ETR, there is no significant change in the PK parameters of DRV. Jose Molto and colleagues evaluated the PK of DRV/cobi, 800mg/100mg once daily, when combined with ETR, 400 mg once daily in HIV-infected persons (abstract O_3). They found ETR substantially reduced cobicistat concentrations with AUC reduced by 30% and trough concentration reduced by about 70%. As would be expected, DRV concentrations were also reduced, most significantly at the trough, which was reduced approximately 40%. Based on these results, it is not recommended to combine DRV/cobi with ETR. If DRV and ETR are clinically necessary, ritonavir should be used as the PK booster, and I would recommend a DRV/r dose of 600mg/100mg twice daily as that is the dose used in the PK and efficacy studies of the combination.
http://regist2.virology-education.com/2017/18AntiviralPK/04_Molto.pdf
3. Dolutegravir (DTG) maximum concentrations (Cmax) are higher in persons more than 60 years, and are associated with a shorter duration of sleep.
In observational cohort studies, up to 5% of HIV-infected persons taking dolutegravir (DTG) have discontinued therapy for CNS adverse effects; this discontinuation rate has been lower in prospective clinical trials; the association between CNS adverse effects and DTG concentrations remains unclear. Dr. Marta Boffito and colleagues conducted an elegant study to evaluate the PK-PD of DTG in HIV-infected persons, 60 years and older, who switched to the FDC of ABC/3TC/DTG. In abstract O_8, they presented data focused on DTG PK and sleep quality. DTG Cmax was higher in this ≥60 year population compared with historical controls <50 years, with geometric mean Cmax values of 4246 vs. 3402 ng/mL. A higher DTG Cmax and AUC were associated with a shorter duration of sleep. There was no association with DTG PK and sleep impairment (insomnia) and no significant change in insomnia sleep scores from baseline (prior to change to DTG) to day 28 after the change. There will be more to come from this study as evaluations continue for 6 months after the change to DTG, and it will be interesting to see whether a longer duration of therapy changes these early results.
Pharm Wk/2017: Dolutegravir Cmax Higher in Elderly, Tied to Shorter Sleep Duration - (06/16/17)
4. Recommendations for statin dosing with cobicistat boosted darunavir (DRV) and atazanavir (ATV).
Abstract O_4 evaluated the PK of rosuvastatin (ROS) and atorvastatin (ATOR) when given with DRV/cobi or ATV/cobi in healthy volunteers. The details of the study design can be found in the slides posted on the conference website. As expected, both DRV/cobi and ATV/cobi caused significant increases in ROS and ATOR concentrations; the increases seen with ATV/cobi on both statins were greater than those with DRV/cobi. The study authors made the following dose recommendations. For DRV/cobi, ROS and ATOR should be initiated at the lowest doses and titrated to response with close monitoring for tolerance/safety. For ATV/cobi, ROS should also be started at the lowest dose and titrated to response with close monitoring for tolerance/safety. For ATV/cobi, they recommended to not exceed an ATOR dose of 10mg once daily. Note, the FDA approved package insert for ATV/cobi makes the same recommendation for ROS: to not exceed a dose of 10mg once daily.
I would be very cautious about using ROS with ATV/cobi, and I would not recommend the combination of ATV/cobi and ATOR. At the 10 mg dose of ATOR used in this PK study, the AUC was increased 9-fold and Cmax was increased 19-fold. This would be equivalent to giving an ATOR dose of approximately 100 mg instead of 10 mg.
Pharm Wk/2017: Evaluation of the Drug-Drug Interaction (DDI) Potential Between Cobicistat-Boosted Protease Inhibitors and Statins - (06/15/17)
C. ARV Pharmacology in Compartments
1. A model of reduced penetration of ARVs into the lymph node predicts virologic failure.
Steven Sanche and colleagues from the University of Montreal and McGill University in Canada gave a fascinating abstract (O_16) modeling the contribution of ARV penetration into the lymph node. They evaluated two hypothesis: one, that all cells have drug concentrations equivalent to those in peripheral blood; and two, that certain physiologic compartments such as the lymph node, have much lower concentrations of ARVs compared with peripheral blood. They found that only the second hypothesis with low lymph node concentrations of ARVs was consistent with observations from clinical trials of virologic success and failure with emergence of resistance.
This paper provides strong motivation for future work on ARV pharmacology in compartments such as the lymph node (and I think the CNS), and for strategies to enhance drug penetration in these compartments.
http://regist2.virology-education.com/2017/18AntiviralPK/25_Sanche.pdf
The Clinical Pharmacology of Therapy for Hepatitis C.
Jennifer J. Kiser, PharmD
A. Drug Interactions with Hepatitis C Therapies and HMG-CoA Reductase Inhibitors ("Statins")
1. Glecaprevir / Pibrentasvir Interactions with Pravastatin, Rosuvastatin, and Dabigatran
Glecaprevir (GLE)/pibrentasvir (PIB) is a fixed-dose direct acting antiviral (DAA) combination likely to receive regulatory approval later this summer. Studies were performed to determine the effects of GLE/PIB on pravastatin, rosuvastatin, and dabigatran concentrations (abstract O_18). These medicines may be used in combination with GLE/PIB in clinical care; additionally, drug interaction studies are often performed with these compounds in order to test the effects of new drugs on cell transporters. Pravastatin is a substrate for the liver uptake transporters OATP1B1 and OATP1B3, rosuvastatin is a substrate for OATP1B1/3 and the efflux transporter BCRP, and dabigatran is a substrate for the efflux transporter P-gp. Results (in table below) indicate that GLE/PIB inhibits OATP1B1/3, BCRP, and P-gp, causing an increase in pravastatin, rosuvastatin, and dabigatran concentrations. Dose adjustments of pravastatin, rosuvastatin, and dabigatran may be required with GLE/PIB.
Effects of Glecaprevir (GLE) / Pibrentasvir (PIB) on the Area-Under-the-Curve (AUC) and Maximum Concentration (Cmax) of Pravastatin, Rosuvastatin, and Dabigatran and Proposed Clinical Management for the Interactions
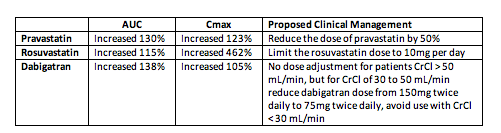
Pharm Wk/2017: DRUG-DRUG INTERACTIONS OF GLECAPREVIR AND PIBRENTASVIR WITH PRAVASTATIN, ROSUVASTATIN, OR DABIGATRAN ETEXILATE - (06/20/17)
2. Sofosbuvir/Velpatasvir with Atorvastatin
Atorvastatin is metabolized by cytochrome P450 3A, and is also a substrate for the drug transporters OATP and P-gp. Velpatasvir inhibits OATP, P-gp and BCRP, so there is a potential for these drugs to interact. When administered with sofosbuvir/velpatasvir, the atorvastatin AUC and Cmax were increased 59% and 68% (abstract P_50). An atorvastatin dose reduction should be considered during sofosbuvir/velpatasvir treatment. Alternatively, the same atorvastatin dose could be maintained with additional monitoring for statin-related side effects.
Evaluation of Drug-Drug Interaction Potential Between Sofosbuvir/Velpatasvir and Atorvastatin - (06/22/17)
B. Drug Interactions with Hepatitis C Therapies and Gastric Acid Modifiers
Ledipasvir is dependent on an acidic pH in the gut for optimal absorption. Thus, the concentrations of ledipasvir can be lower when given with medicines that increase gastric pH such proton pump inhibitors (PPIs). There are conflicting data on whether this reduces the chances of achieving cure of Hepatitis C virus (also known as sustained virologic response (SVR)) with ledipasvir/sofosbuvir (see PMID: 27565882 and PMID: 27533287). In the United Kingdom expanded access program for ledipasvir/sofosbuvir, roughly half of 314 individuals with decompensated cirrhosis were taking a PPI (abstract O_19). After adjusting for time post-dose, body weight, albumin and platelet counts, individuals taking a PPI had ledipasvir concentrations that were 94 ng/mL lower (p<0.0005) than patients not taking a PPI. This is a big difference given the average ledipasvir concentration for all patients in the study was 213 ng/mL. HCV providers were instructed to follow recommendations on PPI use with ledipasvir/sofosbuvir (i.e., given simultaneously with no food and PPI dose not to exceed the equivalent of 20mg of omeprazole once daily), so presumably these were followed. These findings conflict with results from a prior healthy volunteer study (PMID: 27193156), which found minimal effects of 20mg of omeprazole on ledipasvir concentrations. Despite the reductions in ledipasvir concentrations, SVR rates were similar in those taking a PPI (82.4%) compared to those not taking a PPI (84%), p=0.88. All patients were taking ribavirin, which may have offered some protection against the reduced ledipasvir concentrations.
http://regist2.virology-education.com/2017/18AntiviralPK/28_Elsherif.pdf
Grazoprevir (GZR)/ ruzasvir (RZR, MK-8408) / uprifosbuvir (UPR, MK-3682) is a three-drug fixed dose DAA combination in clinical development. Grazoprevir is an NS3/4A protease inhibitor, ruzasvir is an NS5A inhibitor, and uprifosbuvir is an NS5B polymerase inhibitor. Some NS5As inhibitors (ledipasvir and velpatasvir) have pH-dependent absorption, however the AUC of ruzasvir was actually increased by about 25% after 5 days of dosing with omeprazole 40mg once daily (abstract P_41). Grazoprevir AUC was also increased by about 25%. UPR and its major circulating metabolite (M6) were not altered by omeprazole.
C. Drug Interactions with Hepatitis C and HIV Medications
1. DDI with Voxilaprevir (GS-9857) and Antiretroviral Agents
Sofosbuvir (SOF) / velpatasvir (VEL) / voxilaprevir (VOX) is a three-drug fixed dose DAA combination likely to receive regulatory approval later this summer. VOX, an NS3/4A protease inhibitor, is a substrate for cytochrome P450 3A4 and a substrate and inhibitor of P-gp, BCRP, and OATP1B. Thus, there is the potential for clinically significant interactions with ARVs. Data were presented on results of drug interaction studies in healthy volunteers with SOF/VEL/VOX and DRV/RTV/FTC/TDF, EVG/COBI/FTC/TAF, BIC/FTC/TAF, and RPV/FTC/TAF (abstract O_20). As shown in the figure, VOX was increased by DRV/RTV/FTC/TDF and EVG/COBI/FTC/TAF. VOX AUC was increased about 250% and 280% with DRV/RTV/FTC/TDF and EVG/COBI/FTC/TAF, respectively. A greater effect of these combinations on VOX trough was observed (400% and 450%, respectively).
Effect of HIV Antiretroviral Regimens on VOX Pharmacokinetics
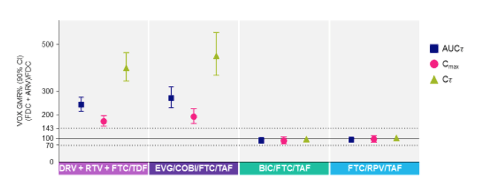
Investigators argued these increases are likely safe based on exposure-safety evaluations in Phase 3 SOF/VEL/VOX studies, but additional data are needed to reassure on the safe use of these ARVs with SOF/VEL/VOX, especially in patients with cirrhosis who have ~73% higher VOX concentrations (abstract P_24). A concern with ledipasvir/sofosbuvir and sofosbuvir/velpatasvir is the increase in tenofovir exposures in those on TDF. The TFV AUC in volunteers taking SOF/VEL/VOX plus DRV/RTV/FTC/TDF was 4600 ng/mL on average. This AUC is within the range of TFV exposures with established renal safety data, which is reassuring on use of the combination. However, additional renal monitoring or avoidance of the combination should be considered in those with impaired renal function. TAF may also be an alternative to TDF in those on ritonavir or cobicistat.
Evaluation of Drug-Drug Interactions Between Sofosbuvir/Velpatasvir/Voxilapevir and Boosted or Unboosted HIV Antiretroviral Regimens - (06/20/17)
2. Doravirine and DAAs
No interaction was observed between doravirine, an investigational NNRTI, and ledipasvir/sofosbuvir (Abstract P_39). Doravirine AUC, Cmax, and C24 were increased 1.56-, 1.41, and 1.61-fold, respectively, with elbasvir/grazoprevir, but this increase is not expected to have clinical relevance (abstract P_42). Doravirine did not alter the pharmacokinetics of elbasvir/ grazoprevir.
Doravirine Does Not Have a Clinically Meaningful Pharmacokinetic Interaction with Ledipasvir/Sofosbuvitr (HARVONI) - (06/20/17)
Doravirine Does Not Have a Clinically Meaningful Pharmacokinetic Interaction with Elbasvir Plus Grazoprevir - (06/20/17)
D. Drug Interactions with Seizure Medications and Sofosbuvir/Daclatasvir
Several seizure medications are contraindicated with DAAs because they could lower DAA concentrations. A reduction in DAA concentrations may reduce the chances of achieving SVR and/or increase the chances of developing viral resistance. Preliminary results from an ongoing study of real-time dose adjustment of daclatasvir/sofosbuvir in patients taking carbamazepine for seizure control were presented (abstract P_36). Daclatasvir doses were increased in all patients to either 60mg twice daily or 60mg thrice daily to account for the reduction in daclatasvir concentrations due to carbamazepine. Even with the dose adjustments, daclatasvir exposures were well below published values. Despite the lower daclatasvir levels, several patients still achieved SVR. Though very preliminary, these data suggest it may be possible to treat patients on seizure medications with DAAs under very controlled settings with real-time dose adjustment of the DAAs.
E. Other Notable Drug Interaction Data:
⋅No interaction was observed between SOF/VEL/VOX and norgestimate/ethinyl estradiol oral contraception (abstract P_40).
⋅Grazoprevir / ruzasvir/ uprifosbuvir does not after the pharmacokinetics of the CYP3A substrate midazolam (abstract P_44).
⋅No clinically significant interaction was observed between dolutegravir and simeprevir (abstract P_48).
Lack of Pharmacokinetic Drug-Drug Interaction Between Norgestimate/Ethinyl Estradiol and Sofosbuvir/Velpatasvir/Voxilaprevir - (06/22/17)
Abbreviations
%CV, percent coefficient of variation
3TC, lamivudine
ABC, abacavir
ACTG, AIDS Clinical Trials Group
ARV, antiretroviral drug
ART, combination antiretroviral therapy
AUC, area under the concentration-time curve
ATV, atazanavir
BCRP, breast cancer resistance protein
CAB, cabotegravir
CAB-LA, long-acting cabotegravir
Cmax, maximum drug concentration
COBI, cobicistat
ConMed, concomitant medication
CrCL, creatinine clearance
CYP, cytochrome P450 drug metabolizing enzymes
DAA, direct acting antiviral agent(s)
DDI, drug-drug interaction
DHHS, Department of Health and Human Services
DTG, dolutegravir
DRV, darunavir
eGFR, estimated glomerular filtration rate
EFV, efavirenz
EU SmPC, European Union Summary of Product Characteristics
ESRD, end-stage renal disease
EVG, elvitegravir
FDA, Food and Drug Administration
FTC, emtricitabine
GALT, gut-associated lymphatic tissue
HCV, Hepatitis C virus
HD, hemodialysis
IM, intramuscular
INSTI, integrase strand transfer inhibitor
MRP2, multidrug resistance protein (efflux transporter)
NVP, nevirapine
OATP, organic anion transporting polypeptide (uptake transporters)
PA-IC90, protein-binding adjusted 90% inhibitory concentration
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamics
P-gp, p-glycoprotein (efflux transporter)
PK, pharmacokinetic
PI, protease inhibitor
PrOD = paritaprevir, ritonavir, ombitasvir, and dasabuvir
RAL, raltegravir
RIF, rifampin
RTV or r, ritonavir
RBV, ribavirin
RPV, rilpivirine
RPV-LA, long-acting rilpivirine
SOF, sofosbuvir
TAF, tenofovir alafenamide
TDF, tenofovir disoproxil fumarate
TFV, tenofovir
TFV-DP, tenofovir diphosphate
Trough, concentration immediately before the next dose
VOX, voxilaprevir (GS-9857)
|
| |
|
|
|
|
|