 |
|
|
| |
CLINICAL PHARMACOLOGY AT THE 12th CROI
|
| |
| |
John G. Gerber, M.D.
Professor of Medicine and Pharmacology
Divisions of Clinical Pharmacology and Infectious Diseases
University of Colorado Health Sciences Center
Denver, Colorado
"understanding of pharmacology during therapy is the only way to optimize efficacy and minimize toxicity of drugs in order to improve clinical outcomes"
"I feel that the drug-drug interaction studies at CROI between omeprazole-ATV and rifampin-ATV have the most important clinical applications"
"The utility of TDM in clinical practice is still controversial"
"Possibly the most compelling evidence against the use of TDM is the lack of adequately powered prospective data demonstrating either efficacy or cost-effectiveness of TDMÉÉ Pregnancy is a situation where TDM can be useful"
"In future studies the effect of gender on drug pharmacokinetics, efficacy, and toxicity should always be at least a secondary goal of the study"
TOPICS:
--TDM
--Atazanavir therapeutic range of drug levels
--Fuzeon & drug levels: Fuzeon may actually be a drug where TDM could be useful
Drug-drug interactions-
--ATC/RTV & omeprazole
--TUBERCULOSIS: ATV & rifampin
--EFV reduced the exposure to buprenorphine
--APV+Kaletra, fosAPV+Kaletra
Drug transporters and the genetic polymorphism to define their roles
--the pharmacogenetics of long term response to EFV and NFV-containing regimen from ACTG 384
-- pharmacokgenetics and NVP hepatotoxicity
-- gene polymorphism in patients receiving IDV, ZDV, and 3TC and correlations to ZDV-TP, 3TC-TP concentrations, and IDV clearance
-- during the third trimester of pregnancy the IDV trough concentrations were ~50% lower: Pregnancy is a situation where TDM can be useful
--gender differences in PK for SQV/RTV & SQV/ATV
The 12th CROI had surprisingly many oral presentations and posters that are of interest to Clinical Pharmacologists specializing in HIV infection. It is impossible to review all the pertinent presentations thus I have picked several specific topics with the presented abstracts that in my opinion affect the clinical approach to patients with HIV infection or topics that are sufficiently controversial that some expert clarification would be helpful. The specific topics include therapeutic drug monitoring (TDM), drug-drug interactions, drug transporters, and some miscellaneous topics that do not neatly fit into any of the other categories.
Therapeutic Drug Monitoring:
The utility of TDM in the therapy of HIV infection is one of the most controversial issues in clinical pharmacology. The European pharmacologists are very pro-TDM while many of the American investigators have a more cautious point of view. In order for TDM to be useful multiple pharmacologic requirements have to be met. These requirements, as well as some of the controversies, were reviewed in recent articles (Gerber and Acosta, The Potential Role of Therapeutic Drug Monitoring in the Treatment of HIV Infection. Topics in HIV Medicine 10:27-32, 2002; Gerber and Acosta, Therapeutic Drug Monitoring in the Treatment of HIV Infection. Journal of Clinical Virology 27:117-28, 2003). Possibly the most compelling evidence against the use of TDM is the lack of adequately powered prospective data demonstrating either efficacy or cost-effectiveness of TDM. With the use of increasingly effective combination therapy and on-treatment HAART efficacy greater than 90% in naï
ve trials, very large clinical trials would be necessary to demonstrate that TDM improves treatment outcomes. TDM may be better utilized to reduce drug-induced toxicity, but again the rate of severe toxicity of commonly used HAART regimens may not be high enough to be able to demonstrate that TDM makes a significant difference. In addition TDM is generally used to monitor a single drug in a three-drug regimen (usually a PI or a NNRTI). This would suggest NRTIs are not as important for either efficacy or toxicity as the other drugs in a regimen, something that has no scientific basis. In addition most practitioners agree that incomplete adherence may be the most common reason for virologic failure of a HAART regimen, something that TDM can overlook especially for short half-life drugs where past indiscretions can easily be missed.
With this background, I want to discuss my take on the posters that were reviewed during Poster Discussion (Session 26) that were ably chaired by Drs. Boffito and Fletcher. Although the idea of an open forum to discuss the strength and weaknesses of these 7 posters was a good one, unfortunately the discussions were generally flat.
Poster 639 by Mentre et al. from France was titled "Prospective Trial to Evaluate How Therapeutic Drug Monitoring of Protease Inhibitors Increase Virological Success and Tolerance of HAART (COPHAR2-ANRS111 Trial)". The title is very misleading because in it there is an assumption that TDM improved the success of PI use, but in reality there was no control group (a group receiving no TDM) thus the authors cannot make those conclusions. The study included 115 PI-naï
ve patients started on a regimen containing IDV/r (n=42), LPV/r (n=38), and NFV (n=35). The study was open-label, non controlled. Trough concentrations were obtained at weeks 2, 8, 16, 24, and 48 and adjustments in dose were made multiple times if necessary before week 24 if concentrations were not within target range of 150-550 ng/mL for IDV, 2500-7000 ng/mL for LPV, and 1500-5500 ng/mL for NFV. Failure was defined as HIV-RNA >200 copies/ml two consecutive times between weeks 16-48 or development of severe toxicity secondary to PI use. Patients without early adverse events were defined as assessable if they had at least the week 16 virological assessment. However as the total group less than 50% of the patients were assessable at week 48 with LPV being the most successful in this regard (25 out of 38). The poster did not account for all the drop-outs. NFV performed the worst with the majority of subjects not achieving therapeutic concentrations and some requiring the use of ritonavir to increase plasma concentration. For LPV many patients required a decrease in dose because the trough concentration of 7000 ng/mL was exceeded. The on-treatment success rate for IDV was 87% and LPV was 81%. The authors concluded that TDM was successfully used in these PI-naï
ve patients. However without a control group it is difficult to conclude that TDM did anything for these patients. For a drug like LPV on-treatment success rate without TDM exceeds 90% and most people with trough concentrations above 7000 ng/mL do not require dose modification. Thus my take on this study is that the authors provide no scientific evidence that the use of TDM did anything to enhance virologic efficacy or reduce toxicity because there was no control group for comparison.
Posters 640 and 641were from the group at UC San Diego and did not truly address the question whether TDM improves the clinical outcome of patients with HIV infection. Poster 640 asked the question whether a population of patients on HAART would benefit from TDM based on expert opinion determination of the correct plasma concentration for that patient. A Bayesian pharmacokinetic modeling was done in real time after an observed drug dose and post dose plasma concentration determinations. The expert committee reviewed the TDM, HIV-RNA, CD4, toxicity and made recommendation regarding the drug therapy. The investigators wanted to define the predictors for the recommendation of change in drug exposure. The patients were randomized to TDM or standard of care. Inclusion was fairly generous and the reason for patient participation was quite varied. A balanced group of patients received NNRTI or PI-based regimens. Out of 177 subjects with week 2 TDM evaluations, 38% had recommendations to alter drug exposure (mostly increase) as determined by the expert TDM panel. The use of LPV, EFV, and greater baseline weight were predictors for the need of TDM. Although this study from Haubrich et al. was elegant in design, it does not attempt to answer the question whether TDM is useful in this mixed population of subjects. Some weaknesses of this analysis include: 1. Only a single drug in the HAART regimen was monitored, 2. Consensus concentration may not be correct despite self-appointed expert panel, 3. The effect of alterations in protein binding and concomitant drugs may make the target concentrations incorrect, 4. Bayesian modeling may come up with the wrong estimate if the assumptions (input) to the model are incorrect. Poster 641 dealt with the development of a computer-based expert system for modeling and interpreting pharmacokinetic data for LPV and EFV. Multiple inputs were outlined in the poster. The computer system performance was compared to the expert committee recommendations. Since there is no gold standard, it is difficult to make any conclusions about which system is correct since no outcomes were measured (at least reported). However EFV post-dose concentrations correlated well between artificial intelligence and expert committee but LPV post-dose concentration was less accurate. Whether in the future this artificial intelligence system will be helpful in improving the clinical outcome of patients on HAART is unknown at this point. The best drugs require minimal monitoring and adherence is still the major impediment to successful therapy. TDM has only a limited utility for successful identification of non-adherence.
Abstract 642 from Johns Hopkins was one of the more interesting studies related to TDM. The original intent of the study was to determine the rate of viral blips in a small group (n=10) of patients where blood was obtained at the same time of the day three times a week for at least 4 months. All subjects had to have plasma HIV-RNA <50 copies/mL on their stable HAART regimen for at least 3 months. The investigators decided to measure the plasma concentrations of the PIs and NNRTIs at the same time as HIV-RNA was obtained, and the time of last dose and adherence was determined by self-report. The expectation was that the variability of plasma concentration will not be very high because these were a motivated group of patients that were willing to show up for blood draws three times a week at the same time of the day. The rate of viral blips were quite low (3.6% in the 713 samples drawn) but the intra-individual coefficient of variation for both PIs and NNRTIs were unexpectedly high. For LPV it varied from 25% to 90% while for EFV it varied from 10% to 60%. There was no ready explanation for this high variation of within patient concentration of these drugs but adherence issues and incorrect recall of the time of the last dose are logical explanations. But these data do beg the question if TDM can be accurately used in a clinic setting where the patients are likely to be less adherent to a regimen than these 10 closely followed patients. Thus making dosage changes after obtaining a single drug concentration for a patient may be inaccurate when the coefficient of variation of plasma concentration drawn at the same time of the day can be as high as 90%.
Poster 643 presented by Dr. Bonora from Torino, Italy examined closely the determinants of virological success in subjects started on an enfuvirtide-based regimen. Complete pharmacokinetics of enfuvirtide was obtained at week 2 and trough concentrations were obtained at weeks 4 and 12. Thirty eight patients were prospectively included in this study. Not surprisingly at week 12 optimized background score (number of active drugs in the regimen) was associated with virological success but also having an ENF Ctrough >2200 ng/mL was independently associated with virological suppression at week 12. The suggestion that ENF therapy could benefit from TDM was considered during the discussion. ENF may actually be a drug where TDM could be useful since administration is by injection and metabolism is by peptidases so there should be less variability in plasma concentration within and across a population. However Dr. Aweeka from San Francisco pointed out that in pediatric patients there was no association between ENF concentration and response. A prospective trial using a randomized design where one group gets dosage modification based on goal plasma concentration while the other group gets fixed ENF dose should be able to answer the question whether TDM is useful during ENF therapy.
Poster 645 presented by Dr. Gonzales de Requena also from Torino, Italy examined trough plasma concentrations of atazanavir (ATV) in 38 patients (66% boosted) from their ATV expanded access program and correlated plasma concentration of ATV to virologic response at week 12. These investigators also collected plasma bilirubin concentration and baseline PI genotypic mutations in order to calculate a GIQ (genotypic inhibitory quotient) as another variable of virologic response. The goal of the study was to establish a therapeutic concentration range for ATV by optimizing the lowest ATV concentration associated with predictable virologic response and upper concentration with acceptable toxicity (bilirubin elevation). In addition calculation of GIQ associated with virologic response could determine the degree of PI resistance that is still associated with a response to ATV. Overall I thought that the study performed on a limited number of patients was rigorously done. The authors found the lower limit of ATV concentration to be 150 ng/mL because 92.6% had virologic response if the trough concentration was >150 ng/mL but only 44.4% response if the ATV Ctrough was <150 ng/mL. The overall virological response rate in this population was 80%. The upper concentration threshold for ATV was determined to be 850 ng/mL because above this concentration 75% of the patients had unconjugated bilirubinemia > 2mg. Since unconjugated bilirubinemia is more of a cosmetic problem than a toxicity problem a more individualized approach to what is an acceptable upper concentration of ATV could be argued. The authors found that GIQ >60 was associated with virologic response. The entire issue of the utility of GIQ requires a prolonged discussion that I do not want to undertake in this review of 2005 CROI. But it is worth commenting that the use of GIQ assumes that all PI genotypic mutations carry equal weight towards resistance something that does not have a rigorous prospective validation.
Drug-Drug Interactions:
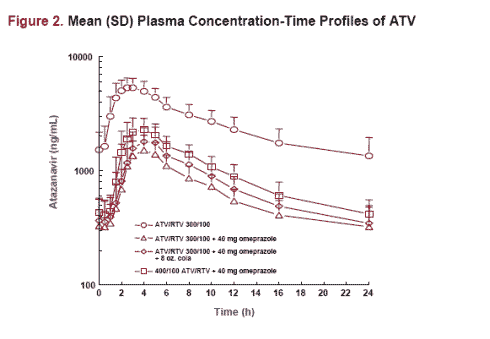
Of the many drug-drug interaction posters/presentations, by far the two with ATV+RTV in healthy volunteers have the greatest clinical implication. ATV/r is gaining momentum as the preferred PI in clinical use secondary to the q.d. dosing and lipid neutral effect. ATV is an organic base with pH dependent solubility. At a low pH the drug is very water soluble something that is necessary for intestinal absorption. As the pH increases the drug has decreased solubility which adversely affects its oral bioavailability. Omeprazole is a potent inhibitor of the gastric acid output by irreversibly inhibiting the H+/K+ ATPase, thus effectively inhibiting acid output for 24 hours. The drug has excellent efficacy for acid-related disorders including severe reflux esophagitis. Omeprazole is available as an OTC drug thus all patients have easy access without input from a physician or a pharmacist. ATV requires the acid milieu of the stomach for optimal solubility and absorption. Thus it would be important to know how a potent gastric acid inhibitor that is commonly used like omeprazole would affect the plasma PK of ATV/r. Poster 658 by Dr. Agarwala examined the effect of omeprazole on ATV PK in HIV-seronegative volunteers. Forty eight subjects participated. All subjects were started on ATV/RTV 300/100 mg q.d. and full PK analysis was performed on day 10. On day 11 the subjects were randomized to three groups (n=16 each). One group had omeprazole 40 mg q.d. added 2 hours prior to ATV/RTV, the other group had omeprazole 40 mg q.d. and ATV/RTV was administered with 8 oz. of cola, and the third group received omeprazole but the dose of ATV/RTV was increased to 400/100 mg q.d.
Omeprazole reduced ATV exposure very significantly. The Cmin decreased by 78% and administering ATV with cola did not help. Increasing ATV from 300 to 400 mg with omeprazole did increase the ATV Cmin but it was still 66% lower than ATV/RTV 300/100 without omeprazole. Omeprazole's effect on RTV PK was minimal.
Based on these data I would not use omeprazole or any proton pump inhibitor with ATV/RTV if high concentration of ATV is the goal. Since omeprazole is available OTC, patients need to be aware that they could be jeopardizing the efficacy of ATV when using concomitant PPIs. How H2 blockers affect the PK of ATV is unknown and until those data are available I would not use H2 blockers with ATV. The impressive data from the poster are in the figure below.
|
|
 |
| |
Abstract 657 dealt with a different mechanism of drug-drug interaction with ATV. In this study, also in HIV-seronegative volunteers, the effect of rifampin on the PK of ATV was evaluated and increasing doses of both RTV and ATV were used to combat the potent inductive effect of rifampin. The poster was presented by Dr. David Burger from the Netherlands. The results of this drug-drug interaction study are of the utmost importance in developing countries where tuberculosis is endemic and rifampin is a critical component of therapy. HIV co-infected subjects need to have access to a potent protease inhibitor-based therapy which at this point is not possible because of the hepatotoxicity associated with high dose RTV with either SQV or LPV and rifampin. The design of this study was elegant. It utilized 72 healthy adults. Eighteen subjects just received 600 mg of rifampin at the end of the study and the data were used as the control for the PK and toxicity of rifampin without concomitant PI. The other 54 subjects received 6 days of ATV 400 mg q.d. with an intensive PK, followed by 10 days of ATV/RTV 300/100 mg q.d. with intensive PK, and then these subjects were randomized to three groups (n=18 each). Group 1 received Rifampin 600 mg q.d. with ATV/RTV 300/100 q.d., group 2 received ATV/RTV 300/200 q.d. with Rifampin 600 mg q.d. for 10 days, and group 3 received ATV/RTV 400/200 with Rifampin 600 mg for 10 days with 24 hour PK performed on the last day. The safety data were relatively encouraging in that grade 2 elevations in ALT/AST occurred in very few subjects (different than what has been reported for LPV/RTV and SQV/RTV). Generally the drug tolerability was excellent. Rifampin was a very potent inducer of ATV metabolism. Rifampin decreased overall exposure of ATV in comparative groups by ~75% but the Cmin decrease was much greater (~95%). The geometric mean Cmin with ATV/RTV 300/100 was 707 ng/mL but decreased to 18 ng/mL with the addition of rifampin. Cmin of ATV increased to 43 ng/mL with ATV/RTV 300/200 and 53 ng/mL with ATV/RTV 400/200 with rifampin. None of these Cmins is adequate for a predictable virologic reponse. The large decrease in Cmin is partly explained by the shortened plasma half-life of ATV with rifampin. The fact that rifampin and ATV/RTV appear safe is encouraging but further studies will be necessary to see if adequate ATV Cmin can be achieved with twice daily dosing of ATV/RTV. These studies are in the planning stages at the ACTG.
Other drug-drug interactions of interest came from Dr. McCance-Katz et al. (poster 653) who found that EFV reduced the exposure to buprenorphine by 50% but no narcotic withdrawal was observed during the 15 days of EFV administration. Clinically buprenorphine is an interesting µ-opioid receptor mixed agonist/antagonist with very high receptor affinity. The drug is metabolized by both conjugation and cytochrome P450 3A4. No doubt the induction of metabolism is secondary to EFV's induction of CYP3A4. Buprenorphine is administered in combination with naloxone in a 4:1 ratio for the treatment of opioid dependence. Although methadone is the drug of choice, EFV is a potent inducer of its metabolism and narcotic withdrawal is well described when EFV is used concomitantly with methadone. Thus finding an opioid agent that can be used safely with EFV would be a major advance in the therapeutics of opioid dependence in HIV-infected patients. In this study despite a 50% reduction in buprenorphine exposure there was no narcotic withdrawal in the 10 subjects. The authors conclude that buprenorphine may be the preferred agent for drug addiction when EFV needs to be used to treat HIV infection. Although this may be the case I have concerns that these subjects were not followed long enough to experience narcotic withdrawal. The long duration of effect of buprenorphine is well described and even after stopping the drug it may take 2 weeks before the onset of narcotic withdrawal. Thus it will be important to collect data on narcotic withdrawal in HIV-infected patients on stable buprenorphine therapy who are started on a long-term EFV-containing regimen.
The concomitant use of Kaletra with APV has been suggested in deep salvage therapy of PI-exposed patients because both LPV and APV may retain some activity despite partial PI resistance. However drug-drug interaction between LPV and APV can result in lowered concentration of both drugs because both APV and LPV are CYP inducers. Paul Pham form Johns Hopkins (abstract 79) presented PK data on APV and LPV from HIV-infected subjects on the combination of APV+Kaletra with or without EFV. A total of 12 subjects were on APV 750 mg + Kaletra 533/133 (LPV/RTV) b.i.d. and 7 subjects received EFV plus APV+Kaletra b.i.d. The study showed that EFV did not affect the PK of LPV or APV although EFV appeared to increase the variability of LPV and APV PK. However it was unclear from the presentation what sort of sample size the study would have needed to demonstrate PK differences secondary to EFV with a large coefficient of variations in the PK of both APV and LPV. However the important point is that APV has been removed from the market and replaced by fosAPV. The drug-drug interaction between fosAPV and LPV is much greater than with APV + LPV as demonstrated in a late breaker poster at the 43rd ICAAC by Angela Kashuba from the ACTG A5143 study. The data she presented demonstrated that concomitant use of fosAPV and Kaletra resulted in ~70% decrease in APV Ctrough and ~60% decrease in LPV Ctrough. Because of this drug-drug interaction the arm of the study using fosAPV+Kaletra was prematurely terminated. Thus it is unclear whether fosAPV should be used with Kaletra. Paul Pham presented some data that fosAPV 1400 mg when combined with Kaletra b.i.d. can give adequate concentrations of both APV and LPV. However median concentrations were presented and not the range so I could not tell what percent of subjects had concentrations outside the acceptable range. I remain dubious whether fosAPV can be used with Kaletra because both LPV and APV concentration variability is quite high and patients harboring partially resistant viruses may not achieve adequate APV and LPV concentrations for a predictable response. Interestingly though Ann Collier from University of Washington presented the efficacy data from ACTG 5143 (poster 577) and did not find that reduction in drug exposure of LPV and APV adversely affected the virological response to the double PI arm of the study. However in A5143 because of the up-front concern about severe drug interaction between fosAPV and Kaletra most of the treated patient harbored APV and LPV susceptible virus. Since the use of double PIs is usually in salvage situation where some level of LPV and APV resistance exists, it is unclear whether adequate concentrations of these PIs can be achieved with fosAPV+Kaletra combination therapy. Until I see positive clinical data I do not use fosAPV in combination with Kaletra in my patients.
Drug transporters and the genetic polymorphism to define their roles:
In the last four years the importance of drug transporters for altering concentrations of HIV protease inhibitors has received significant attention. One aspect of this work has been trying to measure intracellular concentrations of antiretroviral drugs and how the transporters alter this concentration. The importance of this comes from the fact that viral replication occurs within CD4 expressing cells and an accurate estimate of the intracellular concentration of antiretroviral drugs would give a more accurate estimate of their efficacy. However it is important to keep in mind that it is the unbound concentration of drugs that is available for interaction with the virus. At steady state if there were no high affinity active transporters to move drugs unidirectionally the unbound plasma concentration of a drug should be equal to the unbound intracellular concentration. This assumes that the drug can freely traverse membranes. Thus measuring plasma concentration of a drug may be an adequate estimate of the concentration of the drug reaching the cells. However if the cells have a critical number of transporters that can move drugs unidirectionally against a concentration gradient, the plasma unbound concentration and intracellular unbound concentration may not be equivalent. But part and parcel to this question is whether we have any validated tools to measure drug concentrations within cells. The answer appears to be no. Certainly total cellular estimate of a drug may be possible, but it is unclear what fraction of that drug is unbound and available for activity. Thus one might predict that very lipid soluble drugs that tend to bind non-specifically to membranes and other cell components may appear to have high intracellular concentration but in reality the drug may not be available for activity because of the binding. Certainly the data with protease inhibitors tend to confirm this in that lipid soluble drugs like nelfinavir and saquinavir appear to have high intracellular concentration while more water soluble drug like indinavir has low intracellular concentration. However when it comes to antiretroviral activity, indinavir performs every bit as well if not better than nelfinavir or saquinavir.
More recently hypothesis regarding NNRTIs affinity towards cellular transporters have emerged. David Haas from Vanderbilt University had several presentations that suggest that MDR1 (Pgp) polymorphism may be associated with virological failure to EFV (oral abstract 81), and hepatotoxicity to NVP (poster 833). Suggestions that both EFV and NVP are substrates for Pgp came forth but independent confirmation will be necessary since both drugs are lipophilic and move across membranes readily. Presentation of abstract 81 dealt with the pharmacogenetics of long term response to EFV and NFV-containing regimen from ACTG 384. As in ACTG A5097 the authors found that CYP2B6 516 G>T is associated with higher plasma concentration of EFV while CYP 2C19 681 G>A is associated with higher plasma concentration of NFV. Since EFV is metabolized by CYP2B6 and NFV by CYP2C19 the findings were not unexpected or new. However when the genetics associated with virologic response were examined, in subjects receiving EFV (n=340) MDR1 3435TT genotype was associated with decreased virologic failure (similar to what was reported by Fellay in Lancet 2002) but this genotype did not affect the plasma concentration of EFV. Interestingly this association was not seen in patients on NFV despite the fact that NFV has much greater affinity for Pgp. The authors hypothesized (without any data) that decreased Pgp activity may affect intracellular EFV exposure to explain decreased virologic failure. I think that the authors have gone beyond their data by stating this because no definitive evidence has been generated that Pgp affects intracellular EFV movement for this very lipophilic NNRTI. These associations are more hypothesis-generating than definitive evidence. More data with larger sample size need to be generated in order to validate these genetic associations with virologic response.
Poster 833 explored the association between CYP2B6, CYP3A4, CYP3A5, and MDR1 polymorphism and the development of NVP hepatotoxicity in study FTC-302 conducted in South Africa where out of a total of 385 patients receiving NVP, 66 (17%) experienced grade 3 or 4 hepatotoxicity. Of the 66 cases they were able to perform DNA analysis on 53. They also identified 126 controls without hepatotoxicity out of which 108 had adequate DNA for analysis. Interestingly MDR1 3435C>T polymorphism was associated with decreased NVP-induced hepatotoxicity. In this population only 28% expressed the T allele and most of them were heterozygous. No other polymorphism was associated with hepatotoxicity which included the drug metabolizing enzymes. The authors also demonstrated in vitro in a directional cell system that NVP is a weak substrate for Pgp. Again the sample size here was small and I am not convinced that any of these associations are real. More data and a larger sample size need to be studied to validate these findings.
Abstract 650 presented by Andrew Owens from Liverpool University also dealt with the role of transporters on the cellular movement of NVP by measuring intracellular concentration of NVP. These data also tried to correlate plasma concentration of NVP and single nucleotide polymorphisms (SNP) on MDR1, CYP3A4, CYP3A5, and CYP2B6. First the authors found that the intracellular concentration of NVP in PBMCs was very low. They suggested that low NVP cellular concentration is secondary to the lack of expression of OATP-A influx transporter because CEMVBL cells which do express OATP-A have higher cellular NVP concentration which is blocked by an OATP-A inhibitor. This is the first time I have heard that intracellular accumulation of NVP requires an influx transporter. NVP is lipophilic enough to be rapidly absorbed through the GI tract and traverse all membranes including CNS and the placenta. Then finally the authors correlated Pgp expression to intracellular NVP concentrations and found that increased Pgp expression is associated lower intracellular NVP. In addition SNPs in MDR1, CYP3A4, and CYP2B6 were all associated with NVP trough concentrations. It is hard to believe that all these multiple comparisons can be performed in 35 patients for a statistically significant relationship. It is also hard to believe that NVP requires influx transporter for cellular accumulation since this is a lipophilic drug with good diffusion into all tissue compartments. I remain dubious that any of these findings have any relevance.
Anderson from University of Colorado presented poster 649 which examined MRP4, MRP2, and BCRP gene polymorphism in patients receiving IDV, ZDV, and 3TC and correlations of SNPs to ZDV-TP, 3TC-TP concentrations, and IDV clearance were determined. The total number of subjects was 33 but these subjects underwent intensive PK monitoring. The authors found a trend towards faster IDV clearance in subjects with polymorphism in MRP2 promoter after controlling for CYP3A5 expression (previously these authors found that CYP3A5 expression was associated with IDV clearance). The authors also found an association between MRP4 SNP (T4131G) and higher 3TC-TP concentration in PBMCs and MRP4 SNP (G3724A) and higher ZDV-TP concentration. Thus cellular transporter polymorphism may have a role in the variability of the achieved triphosphate concentrations of the nucleoside RTI. The authors correctly conclude that because of the small sample size these data are hypothesis-generating requiring much more additional work.
I hope that the reader got the sense of confusion regarding transporters and HIV drugs. In addition there are dozens of other transporters that have not been examined for relevancy in HIV and therapy and no doubt that SNPs with altered protein activity exist for all these transporters. Transporters without doubt will have a clear role in the movement of some of these drugs across compartments but at this point the field is in its infancy and much more research needs to be performed before scientifically sound and clinically relevant data can be generated. At this point the entire field needs a large dose of skepticism.
Miscellaneous Topics:
Tubiana et al. from Paris presented data (poster 810) on the PK of IDV in pregnant subjects on IDV/RTV 400/100 mg containing regimen. The investigators found that during the third trimester of pregnancy the IDV trough concentrations were ~50% lower (median 236 ng/mL, n=7) as compared to historic data as well as IDV concentration post delivery (440 ng/mL, n=7). This dose of IDV/RTV was well tolerated and resulted in good response. A total of 31 patients completed their pregnancy on IDV/RTV and only 3 experienced detectable viral loads. These three had unusually low plasma concentration of IDV. Intrauterine exposure to IDV did not adversely affect the newborn. Pregnancy is a situation where TDM can be useful since all PIs have lowered concentrations during pregnancy and variable drug exposure during pregnancy may result in virologic failure and development of resistance. TDM can be used to ensure adequate plasma concentration.
Stephen Becker (poster 655) presented PK data on SQV/ATV and SQV/RTV in HIV-seronegative volunteers given these PIs q.d. The most interesting aspect of this the study was the gender differences in the PK of all three PIs. Women consistently achieved at least 2-fold higher concentrations then men corrected for body weight. Gender differences in PI pharmacokinetics have been described previously but the level of difference is much greater in this small cohort of subjects (8 males, 8 females). In future studies the effect of gender on drug pharmacokinetics, efficacy, and toxicity should always be at least a secondary goal of the study. Women experience more toxicity from nucleoside RTI and have increased rate of rash and hepatotoxicity during NVP therapy. As we provide HIV drugs to developing countries where large number of women will be exposed to therapy, it is important to understand gender differences in the pharmacokinetics and safety of antiretroviral drugs.
CONCLUSIONS:
I tried to cover many pharmacology topics from 2005 CROI and I neglected some because of space issues. I feel that I covered the most important and controversial topics. I tried to analyze the data objectively but no doubt personal bias was introduced during data interpretation. I feel that the drug-drug interaction between omeprazole-ATV and rifampin-ATV have the most important clinical applications. The utility of TDM in clinical practice is still controversial but as better and safer drugs are introduced, TDM will become less important in everyday practice. I introduced the abstracts that dealt with drug transporters mainly so the reader begins to understand how uncertain and confusing the field is. However overall understanding of pharmacology during therapy is the only way to optimize efficacy and minimize toxicity of drugs in order to improve clinical outcomes.
|
|
| |
| |
| |
|
|
|
|
|