| |
New insight on hepatitis B virus persistence from the study of intrahepatic viral cccDNA
|
| |
| |
Articles in Press
Journal of Hepatology march 2005
Fabien Zoulim*
INSERM Unit 271, 151 Cours Albert Thomas, 69003 Lyon, France
Article Outline
• 1. Introduction
• 2. What is cccDNA?
• 3. Insight from experimental models
• 3.1. Is cccDNA stable?
• 3.2. Can we prevent the de novo formation of cccDNA?
• 3.3. What is the fate of cccDNA in the liver of infected animals?
• 4. Studies of cccDNA in the liver of chronically infected patients
• 4.1. Detection of intrahepatic cccDNA
• 4.2. CccDNA during the different phases of the disease and insight on the pathobiology of chronic hepatitis B
• 4.3. Evolution of cccDNA in the liver during antiviral therapy
• 5. Perspectives for antiviral therapy
• References
1. Introduction
Chronic hepatitis B virus (HBV) infection remains a major health problem affecting approximately 400 million people worldwide. Although HBV replication is only mildly cytopathic, cellular immune responses directed against the virus can produce substantial liver damage and result in chronic hepatitis, cirrhosis, and hepatocellular carcinoma [1,2]. Chronic hepatitis B (CH-B) encompasses a broad spectrum of disease and can be divided into several phases of natural history which are distinguishable by serological and virological markers. One goal of antiviral therapy is to accelerate patients with chronic active disease into later natural history phases wherein viral replication abates and the incidence of serious liver disease is significantly reduced. With the knowledge gained from the molecular biology of HBV infection, it was shown that HBV covalently closed circular (ccc) DNA plays a key role in viral persistence, viral reactivation after treatment withdrawal, and drug resistance.
2. What is cccDNA?
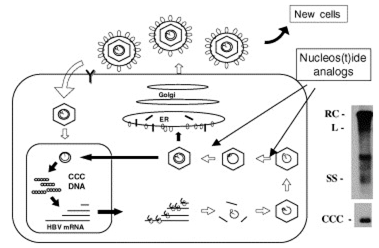
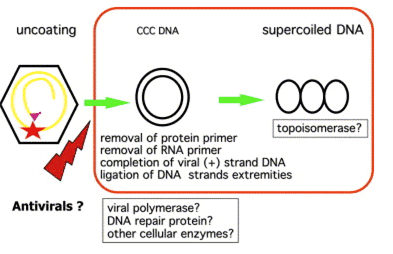
Chronic infection is believed to be maintained by a replicative form of HBV DNA termed covalently closed circular (ccc) DNA (Fig. 1). Formation of cccDNA from incoming virions implies the trafficking of nucleocapsids to the nucleus, the entry of relaxed circular DNA in the nucleus, the completion of plus strand DNA synthesis, the removal of the polymerase primer for minus strand DNA synthesis and of the RNA primer for plus strand DNA synthesis, the ligation of both DNA strand extremities, and its incorporation in the nucleosome to form a non-integrated mini-chromosome 3. During infection, HBV cccDNA accumulates in cell nuclei where it persists as a stable episome and acts as a template for the transcription of viral genes. HBV cccDNA is maintained in the nucleus of infected cells at a level of 30--50 copies per cell [4,5]. It is believed to be methylated to be transcriptionally active. However, the proportion of transcriptionally active copies of cccDNA in individual cells is not known. Viral transcripts are translated into capsid and polymerase proteins and subsequently encapsidated and retro-transcribed into new partially double-stranded viral genomes. DNA-containing nucleocapsids can be enveloped and secreted from the cell as mature virus or cycled back to the nucleus to maintain the cccDNA pool 6. Considering the long half-life of hepatocytes, the limiting factor in eliminating infection is thought to be the clearance of cccDNA reservoirs from infected cells [7,8]. Two immune mechanisms have been proposed to mediate cccDNA clearance: (1) a cytolytic mechanism by which infected cells are eliminated and replaced by cells from an uninfected lineage [9,10] and (2) a non-cytolytic, cytokine-induced 'curing' of infected cells [11,12]. There is evidence from animal models suggesting that both mechanisms are involved and that a concerted action is required to resolve chronic infection in patients. Several important questions remain concerning its half-life and the development of antiviral strategies to prevent its formation or decrease its pool from an already infected liver.
|
|
| |
| |
 |
|
| |
| |
Fig. 1. The HBV replication cycle and the viral DNA replicative intermediates. The left panel shows all the major steps required in the viral replication cycle. Nucleoside(tide) analogs mainly inhibit the viral minus strand DNA synthesis (reverse transcription), and plus strand DNA synthesis (DNA dependent DNA polymerase activity) within viral nucleocapsids. These drugs do not directly affect viral cccDNA which is maintained in the nucleus of infected cells, therefore requiring long-term treatment for a sustained control of viral replication. The right panel shows the analysis of intracellular viral DNA by southern blot hybridization after agarose gel electrophoresis. RC: relaxed circular DNA; DSL: double stranded linear DNA; SS: single stranded DNA; ccc: covalently closed circular DNA.
|
|
| |
| |
 |
|
| |
| |
Fig. 2. Formation of the recalcitrant cccDNA, a difficult target for antiviral therapy Formation of cccDNA from incoming virions implies the trafficking of nucleocapsids to the nucleus, the entry of relaxed circular DNA in the nucleus, the completion of plus strand DNA synthesis, the removal of the polymerase primer for minus strand DNA synthesis and of the RNA primer for plus strand DNA synthesis, the ligation of both DNA strand extremities, and its incorporation in the nucleosome to form a non-integrated mini-chromosome. Currently, none of the viral polymerase inhibitors has been able to prevent de novo cccDNA formation.
3. Insight from experimental models
3.1. Is cccDNA stable?
The importance of cccDNA in HBV persistence at the level of a single cell has been demonstrated in tissue culture experiments. Using nucleoside analogs to block HBV DNA synthesis and recycling of cccDNA by the trafficking of nucleocapsids to the nucleus, it was possible to analyze the half-life of cccDNA in non-dividing cells. Depending on experimental conditions, i.e. using primary duck or woodchuck hepatocytes, or in hepatoma cell lines, a range of results were obtained. Some studies showed that cccDNA is stable in non-dividing hepatocyte cultures with a half-life greater than 30 days 7, other studies showed it has a shorter half-life, while discrepant results suggested that cccDNA is unstable in non-dividing hepatocytes with a half-life of 3--5 days. In the LMH cell line, where hepatocytes are actively dividing, results suggest a shorter half-life. This highlights the difficulty of working with these tissue culture models and different viruses despite belonging to the same family.
In human primary hepatocytes inoculated with HBV, the de novo synthesis of cccDNA and its slow amplification was demonstrated 13. Recently, a new cell line, i.e. HepaRG cells, that is susceptible to HBV infection and support a full replication cycle has been reported 14. This new study model should provide an important tool to study the cellular and viral determinants involved in cccDNA synthesis and its persistence within infected cells. There are, however, neither data available regarding its half-life in human primary cells, nor in this new cell line.
3.2. Can we prevent the de novo formation of cccDNA?
One interesting question is whether nucleoside analogs, by inhibiting the viral DNA dependent DNA polymerase activity, can prevent the de novo formation of cccDNA from the incoming virions. It was shown in primary duck hepatocyte cultures inoculated with DHBV and in Tupaia primary hepatocytes inoculated with HBV, in the presence of lamivudine or adefovir, that these drugs cannot prevent the initial cccDNA formation [15,16]. This has important implications in terms of duration of antiviral therapy, as these results suggest that during antiviral treatment the remaining circulating virions may still have the capacity to infect new cells despite the presence of these drugs.
3.3. What is the fate of cccDNA in the liver of infected animals?
In the past decade, many experiments were performed in in vivo models of hepadnavirus infection including ducks infected with DHBV, woodchucks infected with WHV and chimpanzees infected with human HBV to determine the fate of cccDNA in the liver during the natural clearance of viral infection or during antiviral therapy.
Most experiments in the duck and woodchuck models showed the absence of evidence of a decline of cccDNA per surviving infected cell during antiviral therapy of chronic viral infection.
Although conflicting data emerged in the literature as to the nature of the primary event involved in cccDNA elimination, it was clear that a concerted action of non-cytolytic TH1 immune response exhibiting a direct antiviral effect and cytolytic TH1 cellular response was required during the resolution of an acute infection. Several factors were shown to be essential [8,10--12,17--19]: (1) the production of neutralizing antibodies to prevent infection of new cells by circulating virions; (2) killing of all or some infected hepatocytes and their replacement by non-infected cells (cell regeneration from progenitor cells); (3) hepatocyte proliferation in response to cell killing leading to cccDNA loss after one or more round of cell division; (4) curing of hepatocytes by non-cytolytic TH1 response; (5) block of new cccDNA synthesis within already infected cells.
During antiviral therapy of chronic DHBV or WHV infection, the decline in cccDNA usually precedes the decline in the number of infected cells. Even with the use of potent polymerase inhibitors, cccDNA clearance was not observed explaining the rebound of viral replication after treatment cessation [20,21], and the emergence of drug resistant virus during prolonged therapy 22. The hypothesis is that polymerase inhibitors by inhibiting viral DNA synthesis block the intracellular recycling of nucleocapsids and maintenance of the intranuclear pool of cccDNA. cccDNA is then lost by cell death and dilution through mitosis of hepatocytes.
Available data suggest that cccDNA loss could be explained by cell death 18. Data for cccDNA instability in cell culture may not be reliable because of technical difficulties to distinguish clearance cccDNA versus loss of cccDNA by cell death. However, the results do not rule out mechanisms for cccDNA loss from non-dividing cells. Interestingly, several teams studied the impact of Interferon gamma delivery, a TH1 cytokine, in chronically infected woodchucks to see whether cell curing could occur in these animals. These independent experiments showed the absence of added benefit of the combination of antivirals and IFN gamma compared to nucleoside analog administration alone [23--25].
4. Studies of cccDNA in the liver of chronically infected patients
4.1. Detection of intrahepatic cccDNA
Despite the crucial role of cccDNA during persistent infection and the importance of understanding clearance mechanisms, few data have been collected from patients [26,27]. Indeed, our current understanding of cccDNA has been obtained primarily through studies of the woodchuck and duck hepatitis B virus models [8,9,28,29]. Historical obstacles to the study of HBV cccDNA have been (1) the requirement for liver biopsies, which are difficult to collect, especially from patients in quiescent natural history phases, and (2) the lack of sensitive, specific, and quantitative methods for detection of cccDNA from biopsies.
Notwithstanding these limitations, a few studies were performed to analyze HBV DNA in the liver of infected patients. The presence of cccDNA in the nucleus of infected cells was identified by Southern Blot analysis 26 and its persistence despite inhibition of viral DNA synthesis by antiviral therapy with recombinant leukocyte Interferon was documented 27.
With the development of the PCR technology, there was a need to develop assays to detect and quantify cccDNA in the liver of patients using small size liver tissue samples. Several teams have tried to design PCR methodologies to amplify and quantify more specifically viral cccDNA by choosing primer pairs that preferentially amplify the CCC form and not (or less) the replicative intermediates [30--33]. In a recent study, we have reported the development of a novel real-time PCR assay that allowed us to quantify levels of cccDNA in biopsies collected from CH-B patients 34. This specificity of the methodology is based on the use of two major steps: (1) A plasmid safe DNAse treatment to digest non-covalently close circular DNA, i.e. replicative intermediates. (2) The use of primers located on both sides of the gap of RC DNA to preferentially amplify cccDNA. The quantification of viral DNA was performed by a real time PCR assay using labelled probes and results were normalized to the number of cells by quantifying the beta globin gene.
The specificity of other PCR or non-PCR based (Invader Technology) assays was not demonstrated thoroughly as suggested by the fact that some studies reported levels of cccDNA in serum at levels of 104--106copies/mL. This suggests that these assays may detect RC DNA instead of cccDNA in highly viremic samples 35. It is therefore important to develop standardized assays that could be used on clinical samples for the evaluation of new antivirals undergoing clinical trials.
4.2. CccDNA during the different phases of the disease and insight on the pathobiology of chronic hepatitis B
The cross-sectional natural history study revealed that cccDNA levels were significantly higher in HBeAg+ patients compared to HBeAg-patients, inactive carriers, and patients who underwent HBsAg clearance. This is consistent with the observations of greater levels of intrahepatic HBV replicative intermediates and serum HBV DNA in HBeAg+ patients (compared to the later groups) 36. The observation that cccDNA remained detectable in all HBeAg-patients explains why viral reactivation has been observed, to varying frequencies, in these patient groups. Indeed, fluctuating levels of viral replication and hepatitis are common in patients with precore mutant infection. Viral reactivation and hepatitis flares have also been observed in inactive carriers during long-term natural history studies [37--39]. Even patients with serological evidence of HBV clearance can develop active disease during periods of severe immune suppression such as transplantation, cancer chemotherapy, or steroid use [40,41]. The observed low-level persistence of cccDNA may explain the occurrence of de novo HBV infection in liver transplant patients who received grafts from donors that previously resolved infection. Overall, these observations from the natural history study are consistent with the theory that resolution of hepadnaviral infection occurs by consistent control of viral replication by the host immune system 42.
This is also consistent with the recent description of the case of one patient who failed antiviral therapy with a sequential regimen of entecavir followed by lamivudine, but subsequently cleared HBsAg and seroconverted to anti-HBs after 27 months of adefovir dipivoxil administration added to lamivudine. In this case, viral load declined progressively for the first 19 months of the add-on therapy and then a sharp decline of viremia occurred and was followed by HBsAg clearance and subsequently by the rise in anti-HBsAb titers. None of these events were associated with ALT flares which could have suggested a cytotoxic T cell attack of infected hepatocytes. Analysis of intrahepatic viral DNA showed a sharp decline of total intrahepatic viral DNA consistent with the inhibition of viral DNA synthesis. Furthermore, a significant decline of viral cccDNA was associated with a significant decline of HBV antigen-expressing cells below the detection limit of immunostaining assays 54. Therefore, one may hypothesize that the decline of intrahepatic viral load may be associated in some patients with the sudden restoration of non-cytolytic TH1 response followed by HBs seroconversion 12.
4.3. Evolution of cccDNA in the liver during antiviral therapy
Clinical trials of nucleoside analogs have indicated that short-term therapy is followed by viral recrudescence in most CH-B patients [43,44]. In our study, we showed that antiviral therapy with a potent HBV polymerase inhibitor significantly reduces cccDNA in CH-B patients. Importantly, reductions in cccDNA were correlated with, and similar in magnitude to, reductions in serum HBsAg titer. The parallel change in HBsAg provides further evidence that transcriptionally-active cccDNA is being depleted during therapy. Furthermore, as it is not currently practical to measure cccDNA in biopsies in routine clinical practice, HBsAg quantification in serum may represent a surrogate marker of the intrahepatic cccDNA pool. However, this should be further evaluated.
The rate of cccDNA loss was nearly an order of magnitude lower than that of intracellular HBV replicative intermediates and several orders of magnitude lower than the rate of serum HBV DNA loss. Factors contributing to the relatively slow loss of cccDNA during therapy potentially include (1) the existence of cccDNA as a chromatinized episome (2) the asymmetric nature of hepadnaviral replication which protects cccDNA from being directly depleted by chain-terminating polymerase inhibitors [4,7,28], (3) the preferential use of mature viral nucleocapsids to replenish cccDNA pools during periods of reduced replication [6,7] and (4) the potential for hepatocyte re-infection by residual circulating virus,despite the administration of antivirals as previously shown by tissue culture experiments 15.
These results give insight on the kinetic analyses of viral load decay during potent antiviral therapy. Two main phases of viral load decline have previously been identified: a first phase of rapid viral decline (attributed to the inhibition of viral production in infected cells) and a second phase of slow decline (hypothesized to represent the clearance of infected cells) 45. The slow rate of cccDNA clearance that was observed is likely to contribute to the slower second phase kinetic. The observed longevity of cccDNA also reinforces the prediction that long-term antiviral therapy will be required to control HBV replication unless a vigorous host immune response is mounted 46. However, treatment of CH-B patients with potent nucleos/tides such as lamivudine and adefovir dipivoxil can result in partial restoration of immune responses, at least in some patients, which are necessary for the durable host-mediated control of infection [47,48]. Indeed, since intracellular cccDNA remained detectable in patients that resolved chronic hepatitis B, it may not be necessary to completely eradicate cccDNA to establish long-term control of HBV infection.
Mechanisms for clearance of viral cccDNA have been actively debated. cccDNA clearance during antiviral therapy was not significantly correlated with baseline indicators of host-mediated hepatocyte lysis (e.g. high baseline ALT levels and high baseline histological activity index). This suggests that the decline of cccDNA during antiviral therapy may be occurring primarily through a mechanism other than infected cell killing, i.e. primarily by the potent suppression of viral DNA synthesis which would effectively deplete the pool of mature cytoplasmic nucleocapsids available for conversion into cccDNA.
5. Perspectives for antiviral therapy
The detection of cccDNA in the liver of infected patients may represent an important marker to monitor antiviral therapy. As assays for its quantification are not used routinely, the quantification of cccDNA decline during antiviral therapy may represent an important treatment endpoint in the setting of clinical trials using new antiviral drugs or new combinations. Information gained from such studies may help to provide recommendation for the duration of antiviral therapy and new insight in the mechanism of viral clearance or emergence of drug resistant mutants. The validation of non-invasive surrogate markers such as the quantification of serum HBsAg is required to use this marker in a routine basis. If validated, these markers may help to provide important information to better model the kinetics of viral clearance in patients receiving nulceoside analogs or immune modulators. The development and validation of such assays in the setting of antiviral therapy should open new avenues in the field of anti-HBV therapy in the perspective of adapting treatment strategies to the clinical situation of each infected individual. The discovery of the cellular and virological factors involved in the formation and regulation of cccDNA may also help to identify new molecular targets for antiviral therapy of chronic hepatitis B [49,53].
REFERENCES
1. [1]Lee WM. Hepatitis B virus infection. N Engl J Med. 1997;337:1733--1745. MEDLINE | CrossRef
2. [2]Ganem D, Prince AM. Hepatitis B virus infection—natural history and clinical consequences. N Engl J Med. 2004;350:1118--1129. CrossRef
3. [3]Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51--68. MEDLINE | CrossRef
4. [4]Tuttleman J, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus infected cells. Cell. 1986;47:451--460. MEDLINE | CrossRef
5. [5]Newbold J, Xin H, Tencza M, Sherman G, Dean J, Bowden S, et al.. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol. 1995;69:3350--3357. MEDLINE
6. [6]Wu T-T, Coates L, Aldrich CE, Summers J, Mason WS. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175:255--261. MEDLINE | CrossRef
7. [7]Moraleda G, Saputelli J, Aldrich CE, Averett D, Condreay L, Mason WS. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol. 1997;71:9392--9399. MEDLINE
8. [8]Mason WS, Cullen J, Moraleda G, Saputelli J, Aldrich CE, Miller DS, et al.. Lamivudine therapy of WHV-infected woodchucks. Virology. 1998;245:18--32. MEDLINE | CrossRef
9. [9]Fourel I, Cullen JM, Saputelli J, Aldrich CE, Schaffer P, Averett DR, et al.. Evidence that hepatocyte turnover is required for rapid clearance of Duck Hepatitis B Virus during antiviral therapy of chronically infected ducks. J Virol. 1994;68:8321--8330. MEDLINE
10. [10]Guo JT, Zhou H, Liu C, Aldrich C, Saputelli J, Whitaker T, et al.. Apoptosis and regeneration of hepatocytes during recovery from transient hepadnavirus infections [In Process Citation]. J Virol. 2000;74:1495--1505. MEDLINE | CrossRef
11. [11]Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, et al.. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68--76. MEDLINE | CrossRef
12. [12]Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825--829. MEDLINE | CrossRef
13. [13]Gripon PCD, Guguen-Guillouzo C. Reproducible high level infection of cultured adult human hepatocytes by hepatitis virus: effect of polyethylen glycol on adsorption and penetration. Virology. 1993;192:534--540. MEDLINE | CrossRef
14. [14]Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, et al.. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA. 2002;99:15655--15660. MEDLINE | CrossRef
15. [15]Delmas J, Schorr O, Jamard C, Gibbs C, Trepo C, Hantz O, et al.. Inhibitory effect of adefovir on viral DNA synthesis and covalently closed circular DNA formation in duck hepatitis B virus-infected hepatocytes in vivo and in vitro. Antimicrob Agents Chemother. 2002;46:425--433. MEDLINE | CrossRef
16. [16]Kock J, Baumert TF, Delaney WEt, Blum HE, von Weizsacker F. Inhibitory effect of adefovir and lamivudine on the initiation of hepatitis B virus infection in primary tupaia hepatocytes. Hepatology. 2003;38:1410--1418. MEDLINE | CrossRef
17. [17]Addison WR, Walters KA, Wong WW, Wilson JS, Madej D, Jewell LD, et al.. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J Virol. 2002;76:6356--6363. MEDLINE | CrossRef
18. [18]Summers J, Mason WS. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc Natl Acad Sci USA. 2004;101:638--640. MEDLINE | CrossRef
19. [19]Summers J, Jilbert AR, Yang W, Aldrich CE, Saputelli J, Litwin S, et al.. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc Natl Acad Sci USA. 2003;100:11652--11659. MEDLINE | CrossRef
20. [20]Le Guerhier F, Pichoud C, Guerret S, Chevallier M, Jamard C, Hantz O, et al.. Characterization of the antiviral effect of 2′,3′-dideoxy-2′, 3′- didehydro-beta-L-5-fluorocytidine in the duck hepatitis B virus infection model. Antimicrob Agents Chemother. 2000;44:111--122. MEDLINE
21. [21]Le Guerhier F, Pichoud C, Jamard C, Guerret S, Chevallier M, Peyrol S, et al.. Antiviral activity of beta-L-2′,3′-dideoxy-2′,3′-didehydro-5- fluorocytidine in woodchucks chronically infected with woodchuck hepatitis virus. Antimicrob Agents Chemother. 2001;45:1065--1077. MEDLINE | CrossRef
22. [22]Yamamoto T, Litwin S, Zhou T, Zhu Y, Condreay L, Furman P, et al.. Mutations of the woodchuck hepatitis virus polymerase gene that confer resistance to lamivudine and 2′-fluoro-5-methyl-beta-L-arabinofuranosyluracil. J Virol. 2002;76:1213--1223. MEDLINE
23. [23]Jacquard AC, Nassal M, Pichoud C, Ren S, Schultz U, Guerret S, et al.. Effect of a combination of clevudine and emtricitabine with adenovirus-mediated delivery of gamma interferon in the woodchuck model of hepatitis B virus infection. Antimicrob Agents Chemother. 2004;48:2683--2692. MEDLINE | CrossRef
24. [24]Zhu Y, Cullen JM, Aldrich CE, Saputelli J, Miller D, Seeger C, et al.. Adenovirus-based gene therapy during clevudine treatment of woodchucks chronically infected with woodchuck hepatitis virus. Virology. 2004;327:26--40. MEDLINE | CrossRef
25. [25]Fiedler M, Rodicker F, Salucci V, Lu M, Aurisicchio L, Dahmen U, et al.. Helper-dependent adenoviral vector-mediated delivery of woodchuck-specific genes for alpha interferon (IFN-alpha) and IFN-gamma: IFN-alpha but not IFN-gamma reduces woodchuck hepatitis virus replication in chronic infection in vivo. J Virol. 2004;78:10111--10121. MEDLINE | CrossRef
26. [26]Miller RH, Robinson WS. Hepatitis B virus DNA in nuclear and cytoplasmic fractions of infected human liver. Virology. 1984;137:390--399. MEDLINE | CrossRef
27. [27]Yokosuka O, Omata M, Imazeki F, Okuda K, Summers J. Changes of hepatitis B virus DNA in liver and serum caused by recombinant leukocyte interferon treatment: analysis of intrahepatic replicative hepatitis B virus DNA. Hepatology. 1985;5:728--734. MEDLINE
28. [28]Dandri M, Burda MR, Will H, Petersen J. Increased hepatocyte turnover and inhibition of woodchuck hepatitis B virus replication by adefovir in vitro do not lead to reduction of the closed circular DNA. Hepatology. 2000;32:139--146. MEDLINE | CrossRef
29. [29]Kock J, Schlicht HG. Analysis of the earliest steps of hepadnavirus replication: genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J Virol. 1993;67:4867--4874. MEDLINE
30. [30]Addison WR, Wong WW, Fischer KP, Tyrrell DL. A quantitative competitive PCR assay for the covalently closed circular form of the duck hepatitis B virus. Antiviral Res. 2000;48:27--37. MEDLINE | CrossRef
31. [31]He ML, Wu J, Chen Y, Lin MC, Lau GK, Kung HF. A new and sensitive method for the quantification of HBV cccDNA by real-time PCR. Biochem Biophys Res Commun. 2002;295:1102--1107. MEDLINE | CrossRef
32. [32]Kock J, Theilmann L, Galle P, Schlicht H. Hepatitis B virus nucleic acids associated with human peripheral blood mononuclear cells do not originate from replicating virus. Hepatology. 1996;23:405--413. MEDLINE | CrossRef
33. [33]Mason AL, Xu L, Guo L, Kuhns M, Perillo RP. Molecular basis for persistent hepatitis B virus infection in the liver after clearance of serum hepatitis B surface antigen. Hepatology. 1998;27:1736--1742. MEDLINE | CrossRef
34. [34]Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, et al.. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750--1758. MEDLINE
35. [35]Wong DK, Yuen MF, Yuan H, Sum SS, Hui CK, Hall J, et al.. Quantitation of covalently closed circular hepatitis B virus DNA in chronic hepatitis B patients. Hepatology. 2004;40:727--737. MEDLINE | CrossRef
36. [36]Grandjacques C, Pradat P, Stuyver L, Chevallier M, Chevallier P, Pichoud C, et al.. Rapid detection of genotypes and mutations in the pre-core promoter and the pre-core region of hepatitis B virus genome: correlation with viral persistence and disease severity. J Hepatol. 2000;33:430--439. Abstract | Full Text | PDF (1291 KB) | MEDLINE | CrossRef
37. [37]Davis GL, Hoofnagle JH, Waggoner JG. Spontaneous reactivation of chronic hepatitis B virus infection. Gastroenterology. 1984;86:230--235. MEDLINE
38. [38]Lok AS, Lai CL. Acute exacerbations in Chinese patients with chronic hepatitis B virus (HBV) infection. Incidence, predisposing factors and etiology. J Hepatol. 1990;10:29--34. MEDLINE | CrossRef
39. [39]Loriot MA, Marcellin P, Bismuth E, Martinot-Peignoux M, Boyer N, Degott C, et al.. Demonstration of hepatitis B virus DNA by polymerase chain reaction in the serum and the liver after spontaneous or therapeutically induced HBeAg to anti-HBe or HBsAg to anti-HBs seroconversion in patients with chronic hepatitis B. Hepatology. 1992;15:32--36. MEDLINE
40. [40]Dhedin N, Douvin C, Kuentz M, Saint Marc MF, Reman O, Rieux C, et al.. Reverse seroconversion of hepatitis B after allogeneic bone marrow transplantation: a retrospective study of 37 patients with pretransplant anti-HBs and anti-HBc. Transplantation. 1998;66:616--619. MEDLINE
41. [41]Lau GK, Liang R, Chiu EK, Lee CK, Lam SK. Hepatic events after bone marrow transplantation in patients with hepatitis B infection: a case controlled study. Bone Marrow Transplant. 1997;19:795--799. MEDLINE
42. [42]Rehermann B, Ferrari C, Pasquinelli C, Chisari F. The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nature Medicine. 1996;2:1104--1108. MEDLINE | CrossRef
43. [43]Dienstag JL, Schiff ER, Wright TL, Perrillo RP, Hann HW, Goodman Z, et al.. Lamivudine as initial treatment for chronic hepatitis B in the USA. N Engl J Med. 1999;341:1256--1263. MEDLINE | CrossRef
44. [44]Lai CL, Chine RW, Leung NWY, Chang TT, Guan R, Tai DI, et al.. A one year trial of lamivudine for chronic hepatitis B. N Engl J Med. 1998;339:61--68. MEDLINE | CrossRef
45. [45]Tsiang M, Rooney JF, Toole JJ, Gibbs CS. Biphasic clearance kinetics of hepatitis B virus from patients during adefovir dipivoxil therapy. Hepatology. 1999;29:1863--1869. MEDLINE | CrossRef
46. [46]Lewin SR, Ribeiro RM, Walters T, Lau GK, Bowden S, Locarnini S, et al.. Analysis of hepatitis B viral load decline under potent therapy: Complex decay profiles observed. Hepatology. 2001;34:1012--1020. MEDLINE | CrossRef
47. [47]Boni C, Bertoletti A, Penna A, Cavalli A, Pilli M, Urbani S, et al.. Lamivudine treatment can restore T cell responsiveness in chronic hepatitis B [see comments]. J Clin Invest. 1998;102:968--975. MEDLINE
48. [48]Boni C, Penna A, Ogg GS, Bertoletti A, Pilli M, Cavallo C, et al.. Lamivudine treatment can overcome cytotoxic T-cell hyporesponsiveness in chronic hepatitis B: new perspectives for immune therapy. Hepatology. 2001;33:963--971. MEDLINE | CrossRef
49. [49]Barrasa MI, Guo JT, Saputelli J, Mason WS, Seeger C. Does a cdc2 Kinase-like recognition motif on the core protein of hepadnaviruses regulate assembly and disintegration of capsids?. J Virol. 2001;75:2024--2028. MEDLINE | CrossRef
50. [50]Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol. 2003;77:1885--1893. MEDLINE | CrossRef
51. [51]Summers J, Smith PM, Horwich AL. Hepadnaviral envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 1990;64:2819--2824. MEDLINE
52. [52]Turin F, Bore1 C, Benchaib M, Kay A, Jamard C, Guguen-Guillouzo C, et al. n-Butyrate, a ceI1 cycle blocker, inhibits early amplification of duck hepatitis B virus covalently closed circular DNA after in vitro infection of duck hepatocytes. J Viroal. 1996;70:2691--2696.
53. [53]Borel C, Schorr O, Durand I, Zoulim F, Kay A, Trepo C, et al. Initial amplification of duck hepatitis B virus covalently closed circular DNA after in vitro infection of embryonic duck hepatocytes is increased by cell cycle progression. Hepatology. 2001;34:168--179. MEDLINE | CrossRef
54. [54]Magnard M, Parvaz P, Durantel S, Chevallier M, Chevallier P, Lot M, et al.. Sustained HBS seroconversion during lamivudine and adefovir dipi voxil combination therapy for lamivudine failure. J Hepatol. 2005;42:279--281. Abstract | Full Text | PDF (159 KB) | MEDLINE | CrossRef
|
|
| |
| |
|
|
|