| |
Adiponectin, Rosiglitazone, & Sugar Control; Adiponectin Found to be Protective in Fatty Liver Disease
|
| |
| |
Adiponectin Linked to Sugar Control in Diabetics
NEW YORK (Reuters Heath) - high blood levels of a hormone called adiponectin are associated with improved sugar control in women with diabetes, new research shows.
In addition, high adiponectin levels are associated with high levels of HDL "good" cholesterol and with reduced inflammation. Taken together, these effects could help reduce the risk of heart disease and stroke.
Previous reports have suggested that low adiponectin levels may raise the risk of plaque build-up, or "atherosclerosis," in diabetics. However, the complex interplay between apidonectin levels and various metabolic parameters has not been fully investigated.
Dr. Christos S. mantzoros, from the Harvard Medical School in Boston, and colleagues assessed the association between adiponectin levels and sugar control, cholesterol and inflammation in 925 diabetic women enrolled in the Nurse's health Study.
The researchers' findings appear in the Journal of Clinical Endocrinology and Metabolism.
Adiponectin levels increased as HDL cholesterol levels and physical activity levels rose. By contrast, adiponectin levels dropped as body weight, LDL "bad" cholesterol and various inflammatory proteins increased.
Overall, the results suggest that adiponectin has direct beneficial effects in preventing artherosclerosis, the authors conclude.
Source: Journal of Clinical Endocrinology and Metbalism, August 2005.
Circulating Adiponectin Levels Are Associated with Better Glycemic Control, More Favorable Lipid Profile, and Reduced Inflammation in Women with Type 2 Diabetes
The Journal of Clinical Endocrinology & Metabolism
August 2005 Vol. 90, No. 8 4542-4548
Christos S. Mantzoros, Tricia Li, JoAnn E. Manson, James B. Meigs and Frank B. Hu
Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School (C.S.M.); Departments of Nutrition (T.L., F.B.H.) and Epidemiology (J.E.M., F.B.H.), Harvard School of Public Health; Channing Laboratory (J.E.M., J.B.M., F.B.H.) and Division of Preventive Medicine (J.E.M.), Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts 02215; and General Medicine Division (J.B.M.), Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts 02114
ABSTRACT
Context: Low adiponectin levels, by regulating insulin resistance and metabolic profile, may contribute to the markedly increased risk of atherosclerosis in diabetic subjects.
Objective: The complex interrelationships between adiponectin and metabolic abnormalities have not yet been fully assessed in diabetic women.
Design/Setting/Patients: We performed a cross-sectional evaluation of the association between circulating adiponectin and glycemia, lipid-lipoprotein levels, and inflammatory markers in 925 women with type 2 diabetes enrolled in the Nurses' Health Study.
Results: Circulating adiponectin levels were significantly and positively associated with high-density lipoprotein (HDL) cholesterol and physical activity levels, and inversely with body mass index and plasma concentrations of hemoglobin A1c (HgbA1c), triglycerides, non-HDL cholesterol, apolipoprotein B-100, C-reactive protein, fibrinogen, soluble E-selectin, and soluble intercellular adhesion molecule-1. The above associations were not appreciably altered after adjusting for lifestyle factors, existing medical conditions, obesity, and body fat distribution, with the exception of HgbA1c and soluble intercellular adhesion molecule-1 (which became nonsignificant). Associations between adiponectin and inflammatory markers persisted after control for the potential confounding effects of HgbA1C and HDL cholesterol, suggesting that the antiinflammatory properties of adiponectin are not mediated by its effect on glycemia and lipidemia. With the exception of the associations with triglycerides and apolipoprotein B100, which were significant only in subjects with body mass index less than 30, all other associations observed herein were consistent among obese and nonobese diabetic women.
Conclusions: In summary, higher adiponectin levels are associated with better glycemic control, more favorable lipid profile, and reduced inflammation in diabetic women.
Metabolic effects of rosiglitazone in HIV lipodystrophy: a randomized, controlled trial
Ann Intern Med. 2004 May 18;140(10):786-94.
Hadigan C, Yawetz S, Thomas A, Havers F, Sax PE, Grinspoon S.
Massachusetts General Hospital and Brigham and Women's Hospital, Boston, Massachusetts 02114, USA. chadigan@partners.org
BACKGROUND: Patients with HIV infection who are treated with antiretroviral agents often lose subcutaneous fat and have metabolic abnormalities, including insulin resistance and reduced adiponectin levels, which may be related to disrupted subcutaneous adipogenesis and altered peroxisome proliferator-activated receptor-gamma signaling.
OBJECTIVE: To investigate the effects of rosiglitazone (4 mg/d), a peroxisome proliferator-activated receptor-gamma agonist, in HIV-infected men and women with hyperinsulinemia and lipoatrophy.
DESIGN: A randomized, double-blind, placebo-controlled, 3-month study.
SETTING: University hospital.
PATIENTS: 28 HIV-infected men and women with hyperinsulinemia and lipoatrophy. MEASUREMENTS: Insulin sensitivity measured by euglycemic hyperinsulinemic clamp testing; subcutaneous leg fat area measured by computed tomography; adiponectin, free fatty acid, and lipid levels; and safety variables.
RESULTS: Rosiglitazone, when compared with placebo, improved insulin sensitivity (mean [+/-SD] change, 1.5 +/- 2.1 mg of glucose/kg of lean body mass per minute vs. -0.4 +/- 1.6 mg/kg per minute; P = 0.02), increased adiponectin levels (mean [+/-SD], 2.2 +/- 2.2 micro g/mL vs. 0.1 +/- 1.1 microg/mL; P = 0.006), and reduced free fatty acid levels (mean [+/-SD], -0.09 +/- 0.1 mmol/L vs. 0.01 +/- 0.1 mmol/L; P = 0.02). Mean percentage (+/-SD) of body fat (1.38% +/- 3.03% vs. -0.83% +/- 2.76%; P = 0.03) and subcutaneous leg fat area (2.3 +/- 8.4 cm2 vs. -0.9 +/- 1.9 cm2; P = 0.02) increased significantly with rosiglitazone compared with placebo. Mean total cholesterol levels (+/-SD) also increased with rosiglitazone compared with placebo (0.6 +/- 1.0 mmol/L [25 +/- 37 mg/dL] vs. -0.4 +/- 0.6 mmol/L [-15 +/- 25 mg/dL]; P = 0.007).
LIMITATIONS: The study was relatively small and of short duration.
CONCLUSIONS: The authors demonstrated positive effects of rosiglitazone on lipoatrophy; insulin sensitivity; and metabolic indices, including adiponectin levels, in HIV-infected patients with lipoatrophy and insulin resistance. Peroxisome proliferator-activated receptor-gamma agonists may correct the metabolic abnormalities associated with disrupted adipogenesis in this population. Further studies must determine the clinical utility of such agents in HIV-infected patients.
Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice
Hepatology
Sept 2005
Min You 1 2 *, Robert V. Considine 1, Teresa C. Leone 3, Daniel P. Kelly 3, David W. Crabb 1 2
1Department of Medicine, Indiana University School of Medicine, Indianapolis, IN 2Richard Roudebush Veteran's Affairs Medical Center, Indianapolis, IN 3Departments of Medicine, Molecular Biology and Pharmacology, and Pediatrics, Washington University School of Medicine, Center for Cardiovascular Research, St. Louis, MO
Abstract
The protective effect of dietary saturated fatty acids against the development of alcoholic liver disease has long been known, but the underlying mechanism is not completely understood. We examined the involvement of the adipocyte hormone adiponectin. Circulating adiponectin levels were significantly elevated by chronic ethanol administration to mice consuming a diet high in saturated fat. The increase in circulating adiponectin was associated with the activation a set of hepatic signaling pathways mediated through AMP-activated protein kinase, PPAR-a, and PPAR-° coactivator a, which in turn led to markedly increased rates of fatty acid oxidation, prevention of hepatic steatosis, and alleviation of liver enzyme changes. Furthermore, treatment of rat 3T3-L1 adipocytes with saturated fatty acids (palmitic or stearic acids) in the presence of ethanol increased secretion of adiponectin and enhanced activity of a mouse adiponectin promoter. In conclusion, the protective action of saturated fat against the development of alcoholic fatty liver in mice is partially mediated through induction of adiponectin. The present findings suggest a novel paradigm for dietary fatty acids in the pathogenesis of alcoholic liver disease and provide a promising therapeutic strategy - nutritional modulation of adiponectin - in treating human alcoholic fatty liver disease.
Discussion
The present study provided evidence that adiponectin plays a role in the ability of dietary saturated fat to protect against the development of alcoholic fatty liver in mice. Circulating adiponectin levels were significantly elevated by chronic ethanol administration in the setting of the HSF diet. In parallel, a HSF + E diet stimulated the phosphorylation of AMPK-a and ACC, reduced malonyl CoA contents, and upregulated PGC-1a mRNA and protein levels in the liver. Treatment of rat 3T3-L1 adipocytes with saturated fatty acids in the presence of ethanol increased secretion of adiponectin and enhanced activity of an adiponectin promoter to approximately the same extent. Thus, dietary saturated fat alleviated alcoholic fatty liver partially through upregulation of adiponectin expression and production in adipose tissue. The increased circulating adiponectin is likely to coordinate multiple signaling pathways in the liver, leading to enhanced oxidation of fatty acids, prevention of hepatic steatosis, and alleviation of liver injury.
The precise mechanism through which dietary fatty acids plus ethanol affect adiponectin expression and its secretion has yet to be determined. Adiponectin and TNF-a regulate each other's expression. Downregulation of TNF-a has been proposed as a mechanism for the protective effects of saturated fatty acids.[10][35] Thus, it is possible that the HSF + E diet reduced TNF-a concentrations and thereby enhanced adiponectin expression. Indeed, we found that plasma adiponectin levels were inversely related to serum TNF-a concentrations. However, secretion of adiponectin by 3T3-L1 adipocytes and the response of the adiponectin promoter suggest that dietary fatty acids plus ethanol could also directly regulate adiponectin gene expression. In this scenario, the increased levels of adiponectin in animals fed the HSF + E diet might have led to reduction of TNF-a production. A recent study demonstrated that moderate alcohol consumption significantly enhanced circulating adiponectin concentrations without affecting plasma TNF-a levels, suggesting that regulation of adiponectin by ethanol could be independent of TNF-a levels.[24]
The mouse adiponectin promoter contains a binding site for PPAR-° and its cofactor, retinoid X receptor.[23] Previously, we reported that ethanol treatment slightly but significantly reduced both basal and clofibrate-stimulated PPAR-°-dependent transcriptional activities, and the mRNA and protein levels of retinoid X receptor a were decreased in chronic ethanol-fed mice.[13][14] Therefore, dietary fatty acids plus ethanol may regulate the adiponectin promoter activity through directly modulating PPAR- function.
PGC-1a was initially discovered as a coactivator for PPAR-°. It has subsequently been shown to serve as a coactivator of PPAR-a[33] and is therefore involved in the transcriptional control of the genes encoding enzymes of fatty acid oxidation.[34] Our data suggest a close relationship between PGC-1a expression and the action of adiponectin as demonstrated in cultured hepatoma cells and animal livers. The ability of adiponectin to enhance PPAR-a-responsive reporter activity has been shown in primary hepatocytes.[16] Our studies showed that adiponectin-mediated activation of a PPAR--responsive reporter requires PGC-1 coexpression, indicating that adiponectin may increase the transcriptional activity of PPAR-a through its upregulation of PGC-1a expression. We speculate that PGC-1a may provide a critical signaling link between adiponectin and PPAR-a.
Although our findings strongly suggest that adiponectin acts as an important mediator of the protective action of saturated fatty acids, our data fall short of establishing a cause-effect relationship. However, Xu et al. reported that delivery of recombinant full-length adiponectin into chronic ethanol-fed mice dramatically alleviated fatty liver and alanine aminotransferase abnormalities, partially through adiponectin-mediated induction of hepatic fatty acid oxidation and inhibition of hepatic fatty acid synthesis.[19] In the future, more definitive demonstration of the hypothesis that the protective effect of dietary fatty acids is mediated by adiponectin could be attempted using either adiponectin knockout or hepatic adiponectin receptor 2 null mice.
The possibility that dietary saturated fatty acids plus ethanol directly regulate AMPK, PPAR-a, and PGC-1a cannot be excluded. Saturated fatty acids such as palmitic acid stimulated AMPK activity in the presence of ethanol in cardiomyocytes.[36] We also observed that PGC-1a promoter activity in vitro is modified by various fatty acids only in the presence of ethanol (unpublished data). These direct effects could be, in part, due to the differences in the metabolism of PUFA and HSF in the presence or absence of ethanol.[37] Additional studies addressing these potential multiple mechanisms are required and are currently under investigation in our laboratory.
In conclusion, the translation of our current findings to human beings will need to be studied further. The HSF-rich diet has well-known health risks. The lower PPAR-a expression levels in human liver compared with rodent liver may also limit clinical applications that depend on PPAR-a actions.[38][39] However, our study suggests that nutritional modulation of adiponectin levels may be therapeutic for human alcoholic fatty liver disease.
Adiponectin and alcoholic fatty liver: Is it, after all, about what you eat? EDITORIAL
Hepatology Sept 2005
Frank A. Anania, MD, FACP *
Emory University School of Medicine, Division of Division Digestive Diseases, Atlanta, GA
This is indeed the beginning of a very exciting era in hepatology as we are making rapid progress in understanding how basic metabolism-related hormones are linked with the pathobiology of hepatic steatosis and liver injury. Because hepatic steatosis is critical to the pathophysiology of both nonalcoholic fatty liver disease (NAFLD) and alcoholic fatty liver disease (AFLD); we are on the verge of making substantial breakthroughs in treating two common causes of cirrhosis. The present manuscript by You and colleagues[1] provides compelling data related to the increasing importance of adipokines, or adipocytokines, in fatty liver disease, whether related to alcohol or the metabolic syndrome. Of particular excitement is the clairvoyant explanation of how adiponectin mediates the protective role of saturated fatty acids in AFLD.
Abbreviations:
NAFLD, nonalcoholic fatty liver disease; AFLD, alcoholic fatty liver disease; WAT, white adipose tissue; ALT, alanine aminotransferase; HSC, stellate cell; AMPK, AMP-activated kinase; PPAR, peroxisome proliferators activator receptor; PGC-1a, peroxisome proliferator-activated receptor gamma/coactivator-1; PUFA, polyunsaturated fat; HSF, high-saturated fat; CPT, carnitine palmitoyltransferase; siRNA, small interfering RNA.
Adiponectin, also known as adipocyte complement-related protein or ACRP 30 is a 30 kd protein hormone that is secreted by white adipose tissue (WAT) such as omental fat.[2-4] This adipokine has a plethora of important functions and there is a report that adiponectin has anti-atherogenic potential.[5] Its expression and secretion are dramatically decreased in rodent and human obesity and are lower than age- and sex-matched controls in type II diabetes, insulin resistance, and other conditions associated with metabolic syndrome.[6] In mice, the administration of adiponectin causes glucose-lowering effects and appears to ameliorate insulin resistance. Unlike leptin, there is no evidence of adiponectin resistance in obesity or metabolic syndrome.
Adiponectin may be an attractive therapy for fatty liver disease. Three recent publications provide important background relating the protective effect of adiponectin to various types of liver injury, steatosis, and fibrosis; and these data set the stage for this publication. First, Kamada and colleagues in 2003 demonstrated that liver fibrosis induced by carbon tetrachloride in adiponectin knockout mice was markedly increased compared to that seen in wild-type mice; however, adenoviral expression of adiponectin in the livers of wild-type mice completely prevented carbon tetrachloride-induced fibrosis.[7] Xu and colleagues also recently demonstrated in the ob/ob mouse model of nonalcoholic steatohepatitis that overexpression of adiponectin not only significantly improved clinical features of metabolic syndrome but also significantly improved serum alanine aminotransferase (ALT) and reduced lobular inflammatory activity.[8] Finally, our group has recently shown that adiponectin has the opposite effects on the biological function of activated hepatic stellate cells[9] when compared with leptin.[10] Adiponectin suppresses activated stellate cell (HSC) proliferation, sensitizes HSCs to apoptosis, and may prevent the transitional activation from quiescence.
Given these recent developments, the present series of experiments presented in this issue provide key insights into how adiponectin may be a molecular link between consumption of dietary saturated fatty acids as a defensive shield against AFLD in mice. Dietary fat content and composition have long been implicated in the pathogenesis of alcoholic liver disease; and diets enriched in saturated fatty acids or medium chain triglycerides protect against alcoholic liver injury. Conversely, diets high in polyunsaturated fatty acids promote liver injury.[11][12] On the other hand, the authors point out that chronic ethanol administration is associated with inhibition of AMP-activated kinase (AMPK), and, peroxisome proliferator activator receptor (PPARa). Both signaling molecules are key regulatory elements in hepatic fatty oxidation[13][14] and are the major mediators of the metabolic effect of adiponectin.[15][16] The bulk of the data presented results from a standard feeding model using the Lieber-DeCarli diet.[17] Male C57/BL/6 mice were equally divided into four dietary groups including (1) polyunsaturated fat pair-fed control diet (PUFA); (2) ethanol-containing polyunsaturated fat diet; (3) a high-saturated fat pair-fed control diet (HSF); and (4) an ethanol-containing high-saturated fat diet. Ethanol did not affect liver triglyceride content when added to the HSF diet, there was no increase in serum ALT, and the circulating adiponectin concentration was increased by nearly twofold; conversely, ethanol feeding in the presence of PUFA resulted in a 4.5-fold increase in serum ALT, a significant increase in hepatic triglyceride content, and a fifteen percent decrease in circulating adiponectin. Additionally, an inverse relationship between circulating concentrations of adiponectin and tumor necrosis factor a was also demonstrated; however, the ethanol-treated mice - regardless of dietary fat composition - sustained significant increases in plasma endotoxin compared with the mice which consumed a control diet. This is an important observation because previous reports indicated that alcohol consumption induced endotoxemia and the effects of alcohol were modulated by the fat composition of the diet.[18]
The authors demonstrate that ethanol and HSF - but not PUFA and ethanol - resulted in a robust increase in the phosphorylation of AMPKa - the first signaling element whereby the adiponectin receptor activates its respective transduction cascade. The authors also demonstrate a significant reduction (54%) of malonyl CoA by HSF in the face of ethanol. Malonyl CoA is a potent inhibitor of carnitine palmitoyltransferase I (CPT I), the rate-limiting step for mitochondrial fatty acid oxidation; the PUFA/ethanol diet enhanced malonyl CoA production, the implication of this process results in inhibition of -oxidation of fatty acids. The HSF diet in the ethanol-fed mice increased 3-fold acyl-CoA oxidase messenger RNA. Taken together, the HSF diet in the ethanol-fed mice dramatically increased the capacity for hepatic fatty acid beta-oxidation and the mice handled fatty acid loads more effectively. The remainder of the paper attempts to elucidate key molecular mechanisms to explain the in vivo findings; although the rather extensive cell culture experiments in hepatocytes fall short of showing a clear and direct link between adiponectin and hepatocyte signal transduction. The authors try to link adiponectin to enhanced fatty acid oxidation by various transfection studies including one which demonstrates adiponectin, in a dose-dependent fashion, significantly increased peroxisome proliferator-activated receptor gamma/coactivator-1 (PGCa-1) promoter activity.[19] The rationale for this is clear and is based on the hypothesis that ethanol treatment impairs hepatic fatty acid oxidation by inhibiting the transcriptional activities of PPARa. Adiponectin was shown to increase the reporter activity of PGC-1a in cotransfection experiments; and, PGC-1 is a known coactivator of PPAR-a.[20] Importantly, adiponectin reversed ethanol-associated inhibition of AMPK phosphorylation, increased the rate of palmitic acid oxidation in isolated rat hepatocytes, and adiponectin secretion was increased in 3T3-L1 adipocytes by either exposure to palmitic or stearic acids in the presence of ethanol. The final experiment using HeLa cells indicates that ethanol and palmitate with stearate, but not palmitate and stearate alone, increase adiponectin promoter reporter activity.
The latest work relating adiponectin to AFLD provides a cogent argument whereby saturated fatty acids alleviate AFLD through the upregulation of adiponectin expression. The authors have successfully provided a plausible explanation for previous in vivo observations for a beneficial role adiponectin may play in protection against alcoholic and other types of liver injury. While the present findings are intriguing, this work falls short of a direct link as to exactly how saturated fatty acids - via adiponectin - prevent AFLD. Consequently, the conclusions drawn are speculative. More work on hepatocyte-related adiponectin signaling is now in order to elucidate a causal mechanism. Admittedly, the authors raise these shortcomings, and a compelling paper would have been made even more so if a small interfering RNA (siRNA) for the adiponectin receptor 2 would have been exploited. Because the biology of adiponectin receptors is rapidly developing, such experiments almost certainly will be performed in the near future. Perhaps the HSF diet results in a dramatic increase of adiponectin available to hepatocytes as a result of enhanced production from truncal WAT. The biggest paradox raised in the present work is the danger of human consumption of foods high in saturated fat, as most hepatologists are spending an increasing amount of clinical time these days counseling patients on the dangers of obesity and diabetes. Importantly, modulation of PPARa activity is quite low in humans, and along with the suggestion that nutritional therapy with saturated fatty acids may be beneficial, only underscores how far we have to travel before we can treat AFLD with fat! Perhaps the most promising aspect of this manuscript is that it buttresses the notion that adiponectin is a remedy for both alcoholic and nonalcoholic fatty liver disease and builds on the clear and convincing effects of adipokines in the fundamental biological mechanisms in both hepatocytes and liver nonparenchymal cells. Our current understanding of adiponectin in relationship to hepatocytes is summarized in Fig. 1.
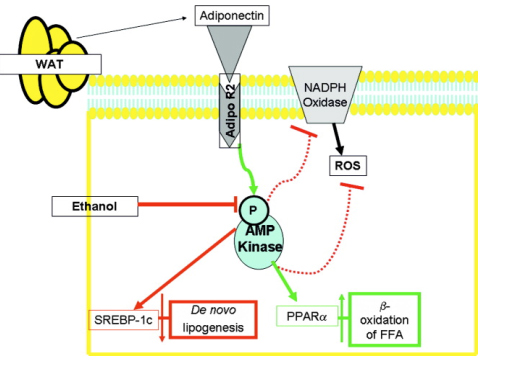
Figure 1. Summary of putative functions of adiponectin in protecting hepatocytes from triglyceride accumulation. White adipose tissue (WAT) secretes adiponectin which via the portal circulation eventually binds to adiponectin receptor R 2 (Adipo R2), the predominant receptor isoform in the liver. AdipoR2 activates AMP Kinase (or AMPK) by phosphorylation which has a multitude of effects including a reduction in pro-oxidant products. By contrast, ethanol inhibits AMPK phosphorylation, which according to the authors, is restored in the presence of ethanol by highly saturated fats (HSF). Importantly, adiponectin-associated AMPK activation may result in a net decrease in hepatocyte triglyceride storage by (1) inhibiting genes (e.g., sterol regulatory element-binding protein-1c, SREBP-1c)[6] required for de novo free fatty acid synthesis, and (2) by increasing the activity of PPAR resulting in increased -oxidation of free fatty acids. In this article, the authors show carnitine palmitoyltransferase I (CPT I) activity, the rate-limiting step for mitochondrial fatty acid oxidation, could be enhanced as is the mRNA for acyl-CoA oxidase in hepatocytes treated with ethanol and the HSF diet. Both are associated with increased fatty acid oxidation.
|
|
| |
| |
|
|
|