| |
Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1
|
| |
| |
Nature Medicine
Advance Online Publication Brief Communication
Published online: 5 October 2005
Gerd Fatkenheuer1, Anton L Pozniak2, Margaret A Johnson3, Andreas Plettenberg4, Schlomo Staszewski5, Andy I M Hoepelman6, Michael S Saag7, Frank D Goebel8, Jurgen K Rockstroh9, Bruce J Dezube10, Tim M Jenkins11, Christine Medhurst11, John F Sullivan11, Caroline Ridgway11, Samantha Abel11, Ian T James11, Mike Youle3 & Elna van der Ryst11
1 Department of Internal Medicine, Division of Infectious Diseases, Haus 11, University of Cologne, Joseph-Stelzmann-Strasse 9, D-50924 Cologne, Germany.
2 Chelsea and Westminster Hospital, 369 Fulham Road, London SW10 974, UK.
3 Royal Free Hospital, Department of Thoracic Medicine, Pond Street, Garrett Anderson Ward, 11th floor, London NW3 2QG, UK.
4 St Georg Hospital, ifi-Institut fur interdisziplinare Infektiologie und Immunologie, Lohmuhlenstr. 5, D-20099 Hamburg, Germany.
5 Klinikum der Johann Wolfgang Goethe-Universitat, Zentrum der Inneren Medizin, Infektionsambulanz, Haus 68, Theodor-Stern-Kai 7, D-60596 Frankfurt, Germany.
6 Head Division Infectious Diseases & AIDS, University Hospital, P.O. Box 85500, 3508 GA Utrecht, The Netherlands.
7 University of Alabama at Birmingham, Birmingham, Alabama 35294-2050, USA.
8 Ludwig-Maximilian-Universitat, Med. Poliklinik der LMU, Munchen, Germany.
9 University of Bonn, Sigmund-Freudstr. 25, D-53105 Bonn, Germany.
10 Beth Israel Deaconess Medical Center, Harvard Medical School, 330 Brookline Ave. CC-913, Boston, Massachusetts 02215, USA.
11 Pfizer Global Research and Development, Sandwich, Kent CT13 9NJ, UK.
The early success of highly active antiretroviral therapy has been tempered by the emergence of cross-resistance, side effects, long-term toxicity and complex dosing regimens1, and there remains a need for well-tolerated, conveniently administered agents with new mechanisms of action. The HIV coreceptor CCR5 is of particular interest as a drug target because a natural mutation (CCR5-Delta32) leads to reduced or absent expression of CCR5 in heterozygous or homozygous genotypes, respectively, with no apparent deleterious consequences. Individuals homozygous with respect to CCR5-Delta32 have a high degree of resistance to HIV infection, whereas heterozygotes have a reduced rate of disease progression2, 3, 4. CCR5 antagonists represent a promising new class of entry inhibitors under development for the treatment of HIV-1 infection5, 6, 7, 8. The CCR5 antagonist maraviroc has potent anti-HIV activity in vitro9 and is safe and well-tolerated at multiple doses up to 300 mg twice daily (BID) in healthy volunteers (M. McHale, S. Abel, D. Russell, J. Gallagher and E. van der Ryst, unpublished results).
We carried out two randomized, placebo-controlled, phase 2a studies (A4001007 and A4001015) to investigate the antiviral efficacy of short-term maraviroc monotherapy in asymptomatic, CCR5-tropic HIV-1-infected patients. Because the study designs (Supplementary Methods online) and patient populations were similar, we combined the results to gain a clearer understanding of the dose-response relationship. Of 163 patients screened, 82 entered the studies. The most common reasons for exclusion were evidence of hepatic impairment (12 of 81), nonphenotypable virus (10 of 81) and a CD4+ cell count of <250 cells/mm3 (10 of 81). Seven patients were excluded owing to detection of dual- or mixed-tropic virus. The overall prevalence of CCR5-tropic virus in screened patients for whom a final tropism result was available was 94% (109 of 116). Patients were randomized to receive 10 d of treatment with maraviroc (25 mg, 100 mg or 300 mg once daily (QD) or 50 mg, 100 mg, 150 mg (fed and fasted) or 300 mg BID) or placebo (Table 1). Baseline demographics and disease characteristics were similar across all treatment groups, with mean baseline viral load and CD4+ counts of 4.62 (range 3.56-5.64) log10 HIV RNA copies/ml and 544 (range 205-1137) cells/mm3, respectively.
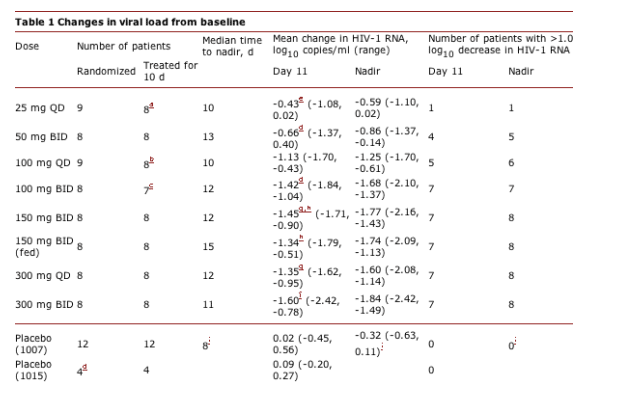
All maraviroc treatment groups had a greater mean decrease in viral load, from baseline to day 11, than did placebo groups (Table 1 and Fig. 1a). Once treatment was discontinued, viral load rebound was not immediate; maximum viral load reduction occurred after day 11 in several patients (median time to nadir = 10-15 d; Table 1). At doses of 100 mg BID and above, all patients, with the exception of one patient with dual- or mixed-tropic virus at baseline (who was inadvertently enrolled owing to a sample-switching error and who experienced no overall reduction in viral load), experienced a reduction in viral load of >1.0 log10 copies/ml at nadir (Table 1). Mean CD4+ count changes between day 1 and day 11 in the maraviroc treatment groups were variable, and there was no correlation between changes in CD4+ count and viral load response or dose (Supplementary Note online).
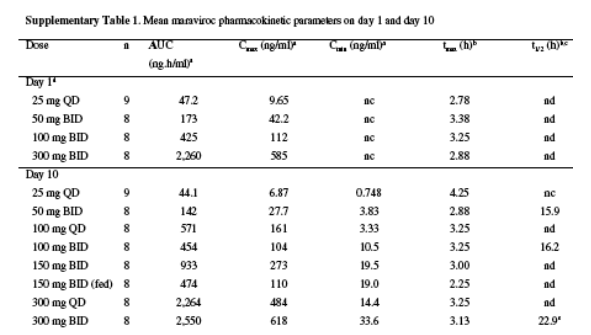
The inconvenience of frequent dosing or food restrictions can represent a barrier to treatment adherence10. We compared BID and QD dosing and the effect of food on response to treatment. There was no difference in reduction in viral load at day 11 between groups treated with 150 mg BID versus 300 mg QD or between groups treated with 150 mg BID that were fed versus fasted. At 150 mg BID, food reduced the Cmax and AUCtau by approx60% and approx50%, respectively, but did not seem to alter the Cmin (Supplementary Table 1 online). Viral load reductions on maraviroc were similar at all doses of 100 mg BID and above, seemed to be close to the maximum on the systemic exposure (AUC) response curve, and were comparable to those observed with potent protease inhibitors and efavirenz11, 12, 13 (Fig. 1b). Taken together, these results imply that maraviroc is likely to be suitable for QD dosing without food restrictions.
Analysis of maraviroc pharmacokinetic data indicated that absorption was rapid but variable, with tmax generally occurring between 1 and 4 h after dosing (Supplementary Table 1). There was no evidence of maraviroc accumulation at any dose when comparing AUC0-12 after single dosing (day 1) with that at steady state after multiple dosing (day 10; Supplementary Table 1). Because maraviroc does not act on a viral target but rather is an antagonist of a human cellular receptor, we also evaluated occupancy of CCR5, determined using an experimental MIP-1beta internalization assay (Supplementary Methods). All maraviroc treatment groups had mean predose CCR5 occupancy of >80% on day 5, and for all doses except 25 mg QD this remained >80% on day 10 predose and >60% on day 15. The delayed rebound in viral load after treatment discontinuation, which has also been described for other CCR5 antagonists14, may be due to prolonged occupancy of CCR5 by maraviroc after treatment had been discontinued. However, no association between degree of CCR5 occupancy and viral load reduction was apparent. A possible explanation for this observation is that very high levels of receptor occupancy are needed for optimal antiviral activity, and that the inherent variability of the experimental assay may result in an inability to detect small differences in receptor saturation. Furthermore, the assay measures only peripheral blood receptor saturation and gives no indication of the degree of tissue receptor occupancy. The relationship between ex vivo receptor occupancy, as measured by this assay, and functional in vivo receptor occupancy is therefore unclear. Finally, the degree to which prolonged CCR5 occupancy delays the rebound in viral load may also be related to the plasma half-life of the drug, as unbound CCR5 molecules are continuously being regenerated in vivo.
Concern exists that selective pressure from a CCR5 antagonist could lead to early predominance of CXCR4-using strains in patients infected with mixed- or dual-tropic HIV. We therefore assessed viral tropism at screening and on days 1, 11 and 40 for all patients. Changes in virus tropism were seen in two patients receiving maraviroc 100 mg QD, both of whom had viral load responses similar to those of other patients in the treatment group. In one patient there was a transient emergence of dual- or mixed-tropic virus on day 11, but on day 40 only CCR5-tropic virus could be detected. In the second patient, dual- or mixed-tropic virus was detected on day 11 and remained detectable at follow-up to day 433. A more detailed description of the analysis of virus tropism in these patients will be published elsewhere (M. Westby et al., unpublished data). These results suggest that CCR5 tropism is strongly selected for in patients, even following the presence of a strong counter-selective pressure. Only long-term studies of CCR5 antagonists, in combination with other antiretroviral drugs, in patients with dual- or mixed-tropic virus will further address this issue.
Maraviroc was well-tolerated at all doses in these studies. The only treatment-emergent adverse events that occurred more than once in any dose group (including placebo) were headache, asthenia, dizziness, gingivitis and nausea (Supplementary Note).
In summary, these results show that the CCR5 receptor is a valid, nonviral target for the treatment of HIV-1-infected individuals and indicate that further clinical evaluation of maraviroc is merited. Similar reductions in viral load have been observed after short-term monotherapy with other investigational drugs in this class14. Given the lack of cross-resistance to other classes and the high prevalence of CCR5 tropic virus in HIV-infected patients, this compound may provide a valuable addition to the armamentarium of antiretroviral drugs.
Supplementary Methods
Study design
Six centers in the UK, Germany and the Netherlands carried out study A4001007 from October 2002 to June 2003. Seven centers in the UK, Germany and the USA carried out
study A4001015 from July to December 2003. We screened Patients 21-40 days prior to
randomization, with the first dose of study drug given 2-7 d later. We randomly assigned
patients to the following treatment groups: maraviroc 25 mg QD, 50 mg, 100 mg or
300 mg BID, or placebo in study A4001007; maraviroc 150 mg BID (fed and fasted), 100
mg or 300 mg QD, or placebo in study A4001015. Patients fasted from midnight to 1 h
(A4001007) or 4 h (A4001015 and A4001007 days 1 and 10) after the morning dose and
were told not to have a large meal within 2 h before or 1 h after the evening dose. Patients
in the fed treatment groups (A4001015 150 mg BID and placebo) took their morning and
evening doses within 30 min of a high-fat (50-60%), high-calorie (1,000 calories)
breakfast or an evening meal supplemented with a standardized high-calorie, high-fat
commercial drink (Ensure Plus_, 500ml), respectively. Patients received treatment for 10 d and were followed up for 30 d after the last dose. Both studies were conducted in
compliance with ethical principles originating from the Declaration of Helsinki, 1989
(Revised Edinburgh October 2000), and with local laws and regulations relevant to the use of new therapeutic agents in the countries of conduct.
Study population
Patients were asymptomatic, HIV-1-infected males or females aged 18-55 years, with
plasma viral load >5,000 HIV RNA copies/ml and CD4 count >250 cells/mm3, who were
either treatment naive or off treatment for ≥8 weeks at the time of screening. Patients with CXCR4- or dual-tropic virus as determined by the ViroLogic Inc. (San Francisco, USA) PhenoSense_ entry assay1 were excluded. Other major exclusion criteria included any clinically significant disease or laboratory abnormality not expected to be associated with HIV infection, AIDS or a previous AIDS diagnosis, and HIV infection diagnosed
<3 months prior to screening or evidence of recent seroconversion.
Efficacy analysis
The primary efficacy end point was the reduction in the log10 plasma HIV-1 RNA load
between baseline and day 11. Secondary efficacy end points were the viral load changes
over the 40-day study period and the time to viral load rebound (defined as the first time
post-randomization that the viral load was greater than or equal to baseline viral load). We assessed viral load at screening, randomization, and on days 1-13, 15, 19, 22, 25 and 40 and was measured by a centralized laboratory (Covance, Geneva, Switzerland) using the Roche Amplicor v1.5 RT-PCR assay (Roche Diagnostics). We calculated the baseline viral load as the mean of the three predose values.
Pharmacokinetic analysis
We took plasma samples for pharmacokinetic assessments pre-morning dose on days 1-10, and at 0, 1, 2, 3 (A4001015 only), 4, 6, 8, 12, 24 h postdose on day 10. In A4001007, we took several additional samples up to 12 h post-morning dose on day 1, as well as 48, 72 and 120 h postdose on day 10. We analyzed maraviroc concentrations at Maxxam
Analytics Inc. (Mississauga, Canada) using high-performance liquid chromatography-
tandem mass spectrometry. We analyzed plasma concentration-time data to determine the minimum and maximum observed plasma concentrations (Cmin and Cmax) and the time to reach Cmax (tmax). We calculated the area under the plasma concentration-time curve (AUC ) from 0_12 h postdose for BID dosing and from 0_24 h postdose for QD dosing. AUC0-24 (2 x AUC_) was also calculated for BID treatment groups. For A4001007, we determined the terminal half-life of the plasma concentration-time curve (t1/2, calculated as ln2/kel, where kel is the apparent terminal elimination phase rate constant) and the accumulation ratio (Rac; AUC0-12 [day 10]/AUC0-12 [day 1]).
Pharmacodynamic analysis
We assessed viral tropism and susceptibility to maraviroc at screening and on days 1, 11
and 40 using the ViroLogic PhenoSense entry assay1. We took samples for the assessment of CCR5 occupancy pre-morning dose on days 1, 5 and 10; 4 h postdose on day 1, and on days 11, 13, 15, 19 and 40. In order to prevent changes in CCR5 expression due to storage and shipping of unprocessed PBMC2, we processed samples according to a standardized experimental MIP-1 internalization protocol at the clinical site within 60 min of collection and fixed in paraformaldehyde. We shipped samples at 2-8 C to a centralized assay laboratory (Esoterix Inc., Austin, USA) within 24 h for flow cytometric assessment. Receptor occupancy was reported as the percentage of cell-surface-expressed CCR5 on peripheral blood lymphocytes (PBLs) that could not be down-regulated when PBLenriched plasma from patients was incubated ex vivo with recombinant MIP-1 . We
measured cell-surface CCR5 expression by flow cytometry using a fluorescently-labeled
CCR5-specific monoclonal antibody (2D7). We calculated percentage receptor occupancy using expression data obtained for PBL aliquots incubated (i) with chemokine in the presence of 1 M maraviroc and (ii) in the absence of additional maraviroc.
We performed CD4+ cell counts in a centralized laboratory (Covance) on samples taken
predose on day 1 and on days 11 and 40 using standard flow cytometry techniques.
Safety and tolerability parameters
We assessed safety and tolerability parameters at each study visit. Safety analyses included physical examinations, laboratory safety tests, and adverse event and serious adverse event recording. The investigator assessed causality. A 12-lead ECG (including measurement of corrected QT interval [QTc]) was carried out and supine and standing blood pressure and pulse rate were measured at multiple time points at screening, pre-morning dose and postdose on days 1, 10 and 40, and pre-morning dose on days 2-9.
Statistical analysis
The primary efficacy analysis population (all evaluable subjects) included all patients
randomized and treated with viral load assessments to at least day 9. The safety analysis
population included all patients who received at least one dose of study drug. We carried
out pharmacokinetic and pharmacodynamic analyses on all patients who had available data for a particular end point.
We used a one-way analysis of variance (ANOVA) to assess differences between
maraviroc treatment groups and the placebo group in the mean reduction in log10 viral load from baseline to day 11. We used the ANOVA output to assess change in viral load using a Williams step-down test in study A4001007. There were no formal statistical analyses performed for the secondary end points. We carried out an ad hoc analysis on data from both trials to determine the maximum viral load decrease over the study period and the time to maximum viral load decrease.
References
1. Coakley, E., Petropoulos, C.J., and Whitcomb, J.M. Assessing chemokine coreceptor
usage in HIV. Curr. Opin. Infect. Dis. 18, 9-15 (2005).
2. Shalekoff, S. & Tiemessen, C.T. Duration of sample storage dramatically alters
expression of the human immunodeficiency virus coreceptors CXCR4 and
CCR5. Clin. Diagn. Lab. Immunol. 8, 432-436 (2001).
Supplementary Note
Mean CD4+ count changes between day 1 and day 11 in the maraviroc treatment groups
were variable and ranged from +5 cells/mm3 (150 mg BID fasted) to +150 cells/mm3
(50 mg BID). In the A4001007 and A4001015 placebo groups mean CD4+ counts
decreased by 2 cells/mm3 and 31 cells/mm3, respectively. There was no correlation between changes in CD4+ count and viral load response or dose.
All adverse events were mild or moderate except for one case of severe diarrhea - in the
150 mg BID (fed) group - which started on day 1 and resolved after 11 d without
pharmacologic intervention. There was no relationship between incidence of any adverse
event and maraviroc dose. There was no clinically significant median change from baseline in any clinical laboratory safety parameter measured for any maraviroc dose or for placebo. Maraviroc had no clinically significant effect on standing or supine systolic or diastolic blood pressure or pulse rate. There was no clinically relevant treatment-related effect on the QTc interval.
|
|
| |
| |
|
|
|